
Abstract
Electrode reactions of intermediates formed during capture of OH radicals by dimethylsulfoxide molecules were studied by laser photoemission in aqueous buffer solutions and pH range from acidic to basic. The results were compared with those obtained previously for electrochemical behaviour of methyl radicals generated via photoemission from CH3Cl. The essential similarity was found for parameters of irreversible one-electron transfer from/to these intermediates, i.e. the potentials E 1/2 on time-resolved voltammograms and rate constants at E = E 1/2. Hence, both active particles were concluded to be equivalent and corresponded to methyl radical. The primary product of OH radicals capture by DMSO molecules, i.e. adduct (CH3)2SO·(OH), was spontaneously decomposed to form ·CH3 with time as low as <2 × 10−5 s. A simultaneous increase of the reduction wave height was observed at pH transition from low basic to low acidic and at illumination times T m of an electrode with UV light if T m ≥ 90–300 ms. The increase exceeded considerably the one-electron reduction level. These features were presumably caused by the rather slow formation of organomercury intermediates as interaction products of the components of the system with a mercury electrode.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The use of optical effects and methods in electrochemical praxis [1–10], e.g., electrochemiluminescence [1–3], photomodulated voltammetry [4, 5] or photoemission [6–9], allows the investigation of reactivity as well as kinetic and thermodynamic characteristics of many intermediates, including those that cannot be created by methods of traditional electrochemistry. Intermediates of electrochemically non-active organic solvents including dimethyl sulfoxide (DMSO) belong to them as well. DMSO is one of the most widespread solvents in electrochemistry [11, 12] due to its universality, high permittivity, chemical and electrochemical stability, low toxicity and other valuable advantages.
However, even being a rather poor acceptor of solvated (hydrated) electron e −s (e −aq ) with respect to other common organic solvents like acetonitrile, dimethyl formamide, propylene carbonate, etc. [13], the rate constant of the electron capture in the case of DMSO is nevertheless of many orders higher than that of water [13]. Such solvents may thus compete for the electron capture with another acceptor added into a solution. Therefore, its concentration has to be relatively high (especially in pure nonaqueous solvents) to prevent the electron capture by a solvent in processes like photoemission/photoinjection (see, e.g., [14, 15]). The reaction products of these solvents with an electron (e −s ) as well as their electrochemical properties are often unknown [13].
Besides the electron capture (reduction), other reactions of solvents with highly reactive radicals are possible. The topic of this study is the reaction of DMSO with ·OH radical which is a strong oxidant.
The reaction of C–S bond rupture at OH radical attack on sulphoxides was first reported by R. Norman et al. in 1964 [16] who evidenced the formation of ·CH3 radical in the system aqueous DMSO–TiIII–H2O2 by ESR technique. The formation of a primary transient OH adduct R2SO·(OH) as a very short-lived intermediate has been suggested, but no direct proofs of its existence was presented so far. An exception is the study [17] where a signal on EPR spectra recorded in photochemical system aqueous DMSO–H2O2 by time-resolved EPR techniques within 100–300 ns was tentatively attributed to this radical. K.-D. Asmus et al. have demonstrated [18] the unimolecular decay of (CH3)2SO·(OH) adduct to ·CH3 and methanesulfinic acid with half-time up to 100 ns. Alkyl radical depletion in this reaction is typical for dialkyl sulfoxides [18].
To study ·OH interaction with DMSO, various ways of OH radical generation were used [16–23], i.e. radiolysis of aqueous systems, the Fenton reaction (Fe2+ + H2O2) or the reaction of hydrogen peroxide with other ions of transition metals (e.g., Ti3+, Cr2+, Cu+, etc.), Haber–Weiss reaction (O2− + H2O2), UV laser (flesh) photolysis, treatment of a solution with oxidase or peroxidase, catalase and other enzymes, etc. A comparative study of reaction of DMSO with OH radicals generated by radiolysis and the Fenton’s reagent was performed in [22]. Not only was the mechanism of the reaction established to be similar but also the yield of products was found to be practically the same. It was also demonstrated that this reaction could have synthetic potentialities [23]. As far as the rate of the reaction of DMSO with OH radicals occurred to be sufficiently high [13], this solvent was proposed to be used as a perspective radioprotector and anti-inflammatory agent for living organisms (see, e.g., [20, 24]).
Since the development of simple and rapid methods of OH radical detection including those in biological systems is still relevant, there is a possibility to use this reaction in voltammetric analysis [25] among other methods for an indirect electrochemical determination of ·OH [26–28].
Methyl radical itself is formed not only by DMSO reaction with OH radical but also with other active species, namely, radical anions CO3·− [21], SO4·− [16, 21] or HO2· radicals [21] although they have somewhat less pronounced redox properties than ·OH [29]. However, according to the opinion of authors [21], it is not desirable to apply DMSO in biological objects as a probe for quantitative and selective indirect detection of OH radicals (via generation of ·CH3) because the HCO −3 ions that are also able to participate in ·OH generation are always present in such systems. The voltammetric [26, 27] or electrochemical impedance [28] detection of OH radicals by their reaction with self-assembled monolayers of alkylthiols on gold electrodes is much more suitable for biological systems.
In the presence of O2, formaldehyde is formed as a main product of the following reaction sequence. DMSO interacts with ·OH, giving rise to the CH3· radicals. They react with O2 to produce CH3OO· radicals [21], which decay rapidly, and the end-products of their transformations are CH3OH and CH2O [21]. All these allowed to call into doubt [21] many statements on non-toxicity of DMSO for living organisms. The voltammetric detection of OH radicals in such cases is carried out on the wave height of formaldehyde reduction [25].
Electrode reactions of ·CH3 were studied previously [19, 30–32]. The radical was generated by the reaction of methane with ·OH at a pressure of 50 atm, and products of pulse radiolysis were investigated by polarography [30]. That radical was obtained also via photoemission capture of e −aq by methyl chloride [31, 32] and studied by photoemission methods.
One more route of ·CH3 generation is reaction of SO4·− radical anion with OAc−, t-BuOH or DMSO, and differential polarograms of products of the reaction were recorded [19]. For unambiguous electrochemical identification of ·CH3 as a product of DMSO reaction with OH radical, it is necessary to study both the reduction and oxidation of this intermediate. The polarographic measurements in [19] have been made, however, only within the range from −1.0 to −1.5 V that was far behind the potentials of ·CH3 oxidation [30, 32]. Thus, the complex information on the electrochemical properties of this system was absent. Therefore, respective measurements for the system DMSO + ·OH within a broad range of experimental conditions (electrode potential, pH, etc.) and determination of the characteristics of electron transfer have to be carried out.
Experimental
A three-electrode cell of quartz was used for laser photoemission experiments. The working electrode was a stationary hanging mercury drop of the Kemula type (see, e.g., [9]). The auxiliary electrode was a Pt foil with a large surface area. All the potentials were referred to a saturated calomel electrode. The range of DMSO concentrations was from 0.03 to 1.3 M. The experiments proceeded in buffer solutions in the pH range 2.4–13.2 with addition of 0.1 M Na2SO4 or 0.5–1 M KCl. The measurements in more acidic solutions were rather difficult due to the competitive capture of e −aq by H3O+ [6, 7]. All the solutions were prepared from triply distilled water. DMSO was purified by a standard procedure like in [9] that included double-distillation under reduced pressure. Dissolved oxygen was removed from aqueous DMSO solutions in photoelectrochemical cell by long-term bubbling with Ar. After that, the solution was saturated with N2O/Ar mixture where N2O concentration was either 0.008 or 0.015 M. N2O was a source of OH radicals generated via the next reaction sequence:
where the rate constant k a of e −s capture by N2O molecule was equal to 6 × 109 M−1 s−1 [6]. N2O and Ar were used without further purification.
Laser photoemission method [6] improved in [8, 9] was used in the measurements of photocurrent J within potentiostatic conditions (the registration of time-resolved voltammograms (TRV) at modulated laser illumination). A photoelectrochemical cell was maintained in a potentiostatic regime with the aid of a specially elaborated high-speed low-current potentiostat. The potential of an electrode E was automatically switched within limits as assigned by an operator. A photocurrent signal from a current–voltage converter was amplified by a preliminary amplifier and supplied on input of a D/A transformer of a computer.
The value of J was obtained by a numerical Fourier transform of a signal from a photoelectrochemical cell illuminated with a modulated light with the period T m = 1.0–10−3 s. Here, T m is the main operation parameter in TRV’s registration like, e.g., scan rate in voltammetry or current density in chronopotentiometry.
Pulse and continuous lasers were used as UV-light irradiation sources. They were nitrogen pulse laser LGI-505 (Russia) (wavelength λ = 337 nm) with a repetition frequency of pulse light of 1 kHz, pulse duration of 8 ns and an average power of ~0.1 W, and continuous He–Cd laser GKKL-8UM(I) (Russia) (λ = 325 nm, an average power of ~0.01 W).
To improve a signal/noise relation, signal registered without illumination was subtracted automatically from the signal accumulated during the electrode illumination. That was the way on how to obtain the dependence of the photocurrent on electrode potential (J, E dependence) for the acceptors investigated. Then, in order to exclude a dependence of the quantum yield of photoemission on the electrode potential, it was normalized to a J 0, E dependence in a solution containing N2O, which is an “ideal acceptor”. The electroreduction of the products of the reaction of N2O with e −aq (OH radicals, cf. Eqs. 1 and 2) occurs at highly positive potentials (the standard potential of OH radical is +2.56 V SCE [29]). Hence, the value of J 0 is proportional to the quantum yield of photoemission over the whole range of the working potential. Thus, it could respect in some degree to a depolarizer reduction within the regime of limiting diffusion current in traditional electrochemistry. A normalized J/J 0, E dependence has the shape of a wave for a radical under study and its height reflects the number of transmitted electrons and therefore does not depend on T m in a general case. The position of the half-wave potential E 1/2 in the axis of potential E is defined by the relation between the rate constants W R/W Ox of reduction/oxidation of adsorbed intermediates R• ads and a period T m of the modulation of the recording signal. The accuracy of the determination of E 1/2 for TRV was equal to ±0.01 V.
This approach allows the determination of absolute values of oxidation and reduction rate constants of intermediates (W ox and W red, respectively) and the creation of the experimental Tafel plots (lg W, E dependencies). The transition from E 1/2, T m dependencies to W, E dependencies is based on the coincidence of the E 1/2 value and the potential where W = KT −1m (K = 5.31 for one-electron irreversible reduction and 10.88 for one-electron irreversible oxidation of R• ads) [8, 9]. A detailed mathematical description could be found elsewhere [7].
In order to expand the effective range of the measured rate constants W R/W Ox, a package of applied programs was developed for the automatic determination of values of E 1/2 and transfer coefficients for electrooxidation β and electroreduction α at higher harmonics of a factual frequency of modulation of illumination of a pulse laser. It provided for the measurement of values of W R/W Ox in the interval 5.0 to 5.0 × 104 s–1. The transfer coefficients were also derived from Tafel plots and TRV’s slope for oxidation and reduction of intermediates [6–8].
Results and discussion
Two well-resolved one-electron waves were observed on TRV’s of intermediates formed during the capture of OH radicals by DMSO molecules (Figs. 1 and 2) within the whole pH range studied. These waves were of approximately equal height that did not depend on T m and pH ~ (6.5–13.2) (Fig. 1) with a slight decrease in more acidic solutions. The E 1/2 of the first wave was within ~−0.30 to −0.56 V, and the E 1/2 of the second one was within ~−1.3 to −1.7 V, respectively, depending on the irradiation time of an electrode with UV light T m.

a Time-resolved voltammograms of solutions containing 0.07 M DMSO and 0.008 M N2O. Dependence on the irradiation time at low basic pH. Britton–Walford buffer +0.1 M Na2SO4, pH 9.1. The irradiation times of an electrode with UV light, T m, ms: 1—1000, 2—30, 3—3.5, 4—0.91, 5—0.3. Dashed line, data of [32] (saturated solution of CH3Cl, phosphate buffer +0.5 M KCl, pH 6.86, T m = 30 ms). b Time-resolved voltammograms of solutions containing 1 M DMSO and 0.008 M N2O. Dependence on the irradiation time at a higher concentration of DMSO and strongly basic pH. Basic buffer +0.1 M Na2SO4, pH 13.2. The irradiation times of an electrode with UV light, T m, ms: 1—1000, 2—290, 3—29, 4—2.9. Dashed line, data of [32] (saturated solution of CH3Cl, Britton–Walford buffer +0.5 M KCl, pH 11.5, T m = 30 ms)

Time-resolved voltammograms of solutions containing DMSO and 0.008 M N2O at different pH values and T m = 30 ms. Buffer solutions with addition of 0.1 M Na2SO4 or 0.5 M KCl. DMSO concentration: 0.07 M (pH 9.1), 0.1 M (pH 7.4), 0.5 M (pH 9.3), 1 M (pH 9.5 and 13.2). N2O concentration: 0.008 M (curves 1, 2, 5), 0.016 M (curves 3, 4). pH: 1 – 7.4, 2 – 9.1, 3 – 9.3, 4 – 9.5, 5 – 13.2. Dashed line – data of [32] (saturated solution of CH3Cl, phosphate buffer +0.5 M KCl, pH 6.86)
Note that the TRV’s for such intermediates were practically coinciding with those recorded by us previously [32] (dotted lines on Fig. 1) within a similar pH range (3.4–12.1) and T m 1.1–10−2 s for methyl radical generated by the reaction:
where k a = 1.9 × 109 M−1 s−1 [13] and the dissociation of the formed radical anion occurred extremely fast and almost barrierless [33].
According to [32], the first wave corresponds to a one-electron irreversible oxidation of ·CH3:
and the second one to its irreversible reduction:
There was a sufficiently broad area between the waves where photocurrents relation J/J 0 was close to 1. Sometimes some distortions (minimums) on TRV’s were observed at the foot of the reduction wave if DMSO concentration was no less than 0.1–0.5 M and pH ≤ 9–10 (Figs. 1 and 2). Such distortions were observed also in [32] and supposed to be associated with adsorption phenomena at CH3· reduction.
Note that although the potential of the reduction wave does not depend on pH like in [19, 30–32], it is valid for oxidation in neutral and low acidic solutions only. The oxidation wave is shifted to more negative values in basic solutions as it is evident from the comparison of TRV’s in Fig. 2 recorded at the same T m (30 ms). According to [30], this may imply to occur in the next process in basic solutions instead of Reaction (4):
The reduction wave height increases both with pH changing from low basic to low acidic values and with T m. This effect took place only for a rather long modulation time T m ≥ 100–300 ms and at a DMSO concentration of more than 0.05–0.1 M. The half-wave potential of this wave is getting more positive at these conditions than that predicted from the Tafel plot for the reduction of ·CH3. The height of the wave considerably exceeded the one-electron level, too (curves 1, 1′–3, 3′ of Fig. 3). It is especially evident from the comparison of curves 1, 1′–3, 3′ and curve 4 from Fig. 3 since the last one corresponds to a one-electron reduction level like all the reduction waves in Figs. 1 and 2. The reduction wave is split into two within the narrow pH range ~6.8–9.1 and T m = 30–900 ms (curves 1 and 2, inset in Fig. 3). The transfer coefficient α can increase here until ~0.65–0.90 (cf. curves 3 and 4, inset in Fig. 3) which is similar to values presented in [31] for the reduction of ·CH3 in 1 M KCl (α ~ 1). Probably, all are caused by the rather slow formation of organomercury intermediates resulting from the interaction of the system components with a mercury electrode. Such phenomena were often observed as processes accompanying adsorption or electrode reactions of organic chalcogenides on mercury [34–36], especially in acidic media and in the presence of halide ions [36]. Organometallic derivatives are able to create a near-electrode condensed phase due to their low solubility in aqueous media [34, 35, 37]. Similar effects were observed previously by photoemission at electroreduction of acetone (in acidic medium) or isopropyl bromide on a lead electrode (see [36, 38]). We did not study these processes in detail.

Time-resolved voltammograms of solutions containing 1 M DMSO and 0.008 or 0.016 M N2O recorded at various pH and T m = 900 (2) or 1000 ms (1, 3) (solid lines) and 300 ms (dashed lines). 1, 1′—0.5 M KCl, 0.016 M N2O; 2, 2′—pH 9.5, 0.016 M N2O; 3, 3′—pH 10, 0.008 M N2O; 4—one-electron level of reduction. T m = 30 ms, pH 10, 0.008 M N2O. Inset, reduction waves in neutral solutions containing DMSO and 0.008 M N2O. DMSO concentration: 0.1 M, pH 7.39 (curves 1, 3, 4), 0.033 M, pH 6.67 (curve 2). T m = 300 ms (curve 1), 30 ms (curves 2, 3), and 3.5 ms (curve 4)
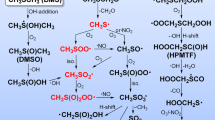
The interaction of OH radicals with DMSO molecules can lead not only to the formation of methyl radicals. According to [18], its mechanism could be described by the following general scheme:

where k OH is within 5.8–7 × 109 M−1 s−1 [13] and for β-fragmentation reaction (Eq. 7b) or (Eq. 7c) k d = 1.5 × 107 s−1 [18].
According to ab initio calculations [39], the fast subsequent decay of (CH3)2SO·(OH) adduct into CH3S(O)OH + CH3· in the gas phase has an energy barrier on −29.8 kJ/mol lower than DMSO + OH interaction although the exothermic formation of one or two products is possible:
Methylsulfinic methyl radical (dimsyl) ·CH2S(O)CH3 has been identified in solution as one of secondary product (Eqs. 7a–7e) reactions [17] with low yield [17, 40] (ca. 5% [40]). It was demonstrated that the route (Eq. 7d) is less favourable in the gas phase compared to the routes (Eqs. 7b and 7c) [39]. Such radicals are usually formed in specific conditions, for example, upon addition of small amounts of sodium methylate [41].
As to the Eq. 7e route, it is the least likely at atmospheric temperatures [39] and no CH3OH was experimentally detected in deaerated solutions [20]. It can be formed in photochemical conditions only in a system DMSO–hydrogen peroxide as a result of secondary reactions (direct interaction of CH3· radical with H2O2 [17]). No other waves except ·CH3 reduction or oxidation were observed on TRV’s. It means that the content of intermediates other than ·CH3 is between 0% and 5%.
Together with Reactions 7b and 7c, the acid-catalysed ionization of (CH3)2S·(OH) is also possible at pH << 6 [42]:
The radical (CH3)2S·(OH) can be tentatively formed as a product of H atom addition generated via the competitive capture of e −aq by H3O+ in sufficiently acidic DMSO solutions (see above). These processes can definitely decrease the concentration of CH3· formed in Reactions 7b and 7c and, consequently, diminish the wave height. Such phenomenon certainly cannot be observed if the radicals were generated via Eq. 3 [32].
The Tafel plots for the reduction of ·CH3 are given in Fig. 4. The dotted line is the corresponding dependence for ·CH3 reduction taken from [32]. From the comparison of Figs. 2 and 4, one can conclude that the reduction rate of ·CH3 radical practically does not depend on pH within the range from 3.4 to 13.2. A small shift to a more negative potential of ca. 0.05–0.07 V is only observed in basic solutions at pH ~10–13. The absence of such dependence, especially in acidic solutions, confirms a direct electron transfer from electrode to methyl radical. There is no appreciable dependence of the reduction rate neither on the nature and concentration of a background electrolyte (0.1–1 M KCl, Na2SO4) nor on the composition of a buffer. The Tafel plots for the reduction of “both” types of ·CH3 radicals, i.e. generated either from CH3Cl [32] or from DMSO (this study), as well as their transfer coefficients α (0.49 and 0.51 (±0.05), respectively) are sufficiently close.

Tafel plots for ·CH3 oxidation rate constants W ox (left side) and reduction rate constants W red (right side). The dashed line and filled symbols are data of [32]
The values of E 1/2 for the one-electron reduction of methyl radical reported by other authors were as follows: −1.43 V (1 M KCl, photoemission) [31], −1.42 V (pH 3–11.5, buffer solutions of 0.3 M Na2SO4 with addition of HClO4, H2SO4 or NaOH, polarography of products of pulse radiolysis) [30] and −1.33 V (buffer solutions of 0.06 M KH2PO4–0.01 M Na2B4O7, pH 5.8–9.0, differential polarography; the potential scan rate was 0.25 V/s) [19]. One can see that all these values fall into the potential range of the Tafel plot in Fig. 4 derived in this study, ca. −1.25 to −1.70 V. However, there are problems to compare these potentials obtained by various methods [7]. The main reason is that, in the mentioned investigations [19, 30, 31], no attempts were made to determine the rate constants of ·CH3 reduction at the respective potentials.
Some authors [19] claim, however, that the process of ·CH3 reduction is reversible, which is in disagreement with the present study as well as with [32]. According to [6, 9], the irreversibility of alkyl radicals reduction including ·CH3 is caused first of all by the very low stability of respective carbanions in protic media, even in spite of a sufficiently high barrier of protonation of alkyl carbanions (ca. 150–200 kJ M−1) [6]. The standard potential E 0 of ·CH3/CH −3 redox couple was estimated to be −1.01 ± 0.16 V [6] (calculated from the Tafel plot for ·CH3 reduction [32] and the thermodynamic cycle: CH4 (g) → CH4 (aq) → CH −3 (aq) → ·CH3 (aq) → ·CH3 (g) [6, 7]), and −1.19 ± 0.15 V [43] (obtained by a method of indirect reduction, namely, competition between coupling and reduction of radicals by radical anions [44]). Thus, the difference between E 0 and E 1/2 for ·CH3 reduction can reach 0.3–0.7 V. This difference should be attributed to an overvoltage caused by the slow ·CH3 reduction (Eq. 5).
The W Ox, E dependencies for oxidation of the radical within pH range from low acidic to strongly basic are also presented on the Fig. 4. In distinction from the reduction, the oxidation rate of ·CH3 depends on pH in basic solutions but such dependence practically does not take place in neutral and low acidic media. The respective shift of E 1/2 from neutral to strongly basic solutions (i.e. from pH 7.1–7.5 to 13.2) consists of ca. 0.25–0.3 V that is in qualitative agreement with the data of [30] where E 1/2 values changed from ca. −0.18 to −0.22 V (pH 4–6) to ca. −0.42 V (pH 11.5). The transfer coefficient β for oxidation occurred to be too high and apparently did not correspond to a simple electron transfer. Together with available dependence of ·CH3 oxidation rate on pH, it could represent an influence of a certain chemical stage on the electrode process (for example, an antecedent formation of a metastable complex of an intermediate with a proton acceptor [45]).
The Tafel plots for reduction are straight lines within a sufficiently broad range of measured W R. It means that the electrode process corresponds to a simple one-electron transfer that is not complicated with chemical steps. No deviations are present on the respective plot for ·CH3 reduction generated from DMSO until W R = W max ~ 5 × 105 s−1. Hence, the lifetime of (CH3)2SO·(OH) adduct is <1/W max, i.e. it is <2 × 10−5 s.
Conclusions
Electrode reactions of intermediates formed during the capture of OH radicals by dimethylsulfoxide molecules according to the known reaction scheme (Eq. 7) were studied by laser photoemission. The results of the present study were compared with those from [32] where ·CH3 radicals were generated via photoemission from CH3Cl. It was concluded that the same radical was formed in the reaction of OH radicals with DMSO molecules. According to the scheme (Eq. 7), it should be considered as one of the secondary decay products of short-lived intermediate (CH3)2SO·(OH) which lifetime was found to be <2 × 10−5 s. The similarity was revealed for electrochemical characteristics of one-electron irreversible reduction and oxidation of ·CH3 radicals generated by the above mentioned different ways (potentials E 1/2 of time-resolved voltammograms and rate constants of their electrode reactions).
Several possible routes of formation of various intermediates in the reaction of OH radicals with DMSO were considered but no signals other than ·CH3 reduction or oxidation were detected. However, the increase of the reduction wave height both with pH changing from basic to acidic values and with T m was observed. This effect took place only at rather long illumination time T m ≥ 100–300 ms and at a DMSO concentration higher than 0.05–0.1 M. The reduction wave height considerably exceeded the one-electron level under these conditions. Its half-wave potential was shifted more positively than that predicted from the Tafel plot for the reduction of ·CH3. All these phenomena are most probably caused by the rather slow formation of organomercury intermediates resulting from the interaction of the components of the system with a mercury electrode.
The reduction rate of ·CH3 did not depend on pH and corresponded to a simple one-electron transfer. On the other hand, the rate of ·CH3 oxidation has been found to depend on pH in basic media, which indicated a possible participation of OH- in the process. The presented data are in a good agreement with those available in literature, where ·CH3 was generated by alternative ways. The findings described represent an electrochemical evidence of the formation of methyl radicals as a result of the capture of OH radicals by DMSO molecules.
Hence, DMSO should not be used as a solvent in electrochemical investigations of systems where active oxidants like OH radicals or other highly reactive agents of similar properties are present, unless generation of methyl radicals is needed.
References
Ludvik J, Pragst F, Volke J (1984) Electrochemical generation of triplet states: simplified estimation of triplet energies by electrogenerated chemiluminescence based on the anodic cleavage of dimeric dihydroheteroarenes. J Electroanal Chem 180(1–2):141
Ludvik J, Volke J, Pragst F (1986) Investigation of 2 radical intermediates in the anodic-oxidation of 1,4-dihydropyridines by electrochemiluminescence. J Electroanal Chem 215(1–2):179
Ludvik J (1994) Elektrochemicky generovaná luminescence (in Czech). Chem Listy 88(11):696
Wayner DDM, Parker VD (1993) Bond-energies in solution from electrode-potentials and thermochemical cycles—a simplified and general-approach. Acc Chem Res 26(5):287
Grampp G, Muresanu C, Landgraf S (2005) Solvent influence on the electrochemical reduction of photochemically generated cis-azobenzene. J Electroanal Chem 582(1–2):171
Krivenko AG, Benderskii VA (1990) Electrochemistry of short-lived intermediates. Russ Chem Rev 59(1):1
Benderskii VA, Benderskii AV (1995) Laser electrochemistry of intermediates. CRC, New York
Krivenko AG, Kotkin AS, Kurmaz VA (2005) Thermodynamic and kinetic characteristics of intermediates of electrode reactions: determination by direct and combined electrochemical methods. Russ J Electrochem 41(2):122
Krivenko AG, Kotkin AS, Kurmaz VA (2005) Thermodynamic and kinetic characteristics of intermediates of electrode reactions: a comparative investigation of a number of alkylaryl and alkyl halide radicals by the laser photoemission methods. Russ J Electrochem 41(2):137
Gründler P, Kirbs A, Dunsch L (2009) Modern thermoelectrochemistry. ChemPhysChem 10(11):1722
Lund H (1983) Practical problems in electrochemistry. In: Lund H, Baizer MM (eds) Organic electrochemistry. An introduction and a guide, 2nd edn. Marcel Dekker, New York, pp 161–233
Gritzner G (2011) Half a century of electrochemistry, mainly in non-aqueous electrolytes (a personal view). J Solid State Electrochem 15 (7/8) 1791
Buxton GV, Greenstock CL, Helman WPh, Ross AR (1988) Critical-review of rate constants for reactions of hydrated electrons, hydrogen-atoms and hydroxyl radicals (.OH/.O−) in aqueous-solution. J Phys Chem Ref Data 17(2):513
Hapiot Ph, Konovalov VV, Savéant J-M (1995) Application of laser pulse photoinjection of electrons from metal electrodes to the determination of reduction potentials of organic radicals in aprotic solvents. J Am Chem Soc 117(4):1428
Krivenko AG, Kotkin AS, Kurmaz VA (2002) Prospects for determination of thermodynamic and kinetic parameters of intermediates of electrode reactions by laser photoemission. Mendeleev Commun 12(1):11
Dixon WT, Norman ROC, Buley AJ (1964) Electron spin resonance studies of oxidation. 2. Aliphatic acids + substituted acids. J Chem Soc 3625
Woodward JR, Lin TS, Sakaguchi Y, Hayashi H (2000) Detection of transient intermediates in the photochemical reaction of hydrogen peroxide with dimethyl sulfoxide by time-resolved EPR techniques. J Phys Chem A 104(3):557
Veltwisch D, Janata E, Asmus K-D (1980) Primary processes in the reaction of OH·-radicals with sulphoxides. J Chem Soc Perkin Trans 2(1):146
Song JF, Chen ZP, Guo W, Wang FM (2003) In situ generation and detection of methyl radical by voltammetry. Chin Sci Bull 48(11):1093
Koulkes-Pujo AM, Moreau M, Sutton J (1981) Methane formation from the reactions of hydroxyl radicals and hydrogen-atoms with dimethylsulfoxide (DMSO). FEBS Lett 129(1):52
Herscu-Kluska R, Masarwa A, Saphier M, Cohen H, Meyerstein D (2008) Mechanism of the reaction of radicals with peroxides and dimethyl sulfoxide in aqueous solution. Chem Eur J 14(19):5880
Eberhardt MK, Colina R (1988) The reaction of OH radicals with dimethyl sulfoxide. A comparative study of Fenton’s reagent and the radiolysis of aqueous dimethyl sulfoxide solutions. J Org Chem 53(5):1071
Bertilsson B-M, Gustafsson B, Kühn I, Torssel K (1970) Generation of radicals from sulphoxides with Fentons reagent—new radical alkylation method. Acta Chem Scand 24(10):3590
Raju MR, Schillaci ME, Carpenter SG, Goodhead DT, Ward JF (1996) Radiobiology of ultrasoft X rays. V. Modification of cell inactivation by dimethyl sulfoxide. Radiat Res 145(5):563
Zou H, Tai C, Gu XX, Zhu RH, Guo QH (2002) A new simple and rapid electrochemical method for the determination of hydroxyl radical generated by Fenton reaction and its application. Anal Bioanal Chem 373(1–2):111
Scholz F, Gonzalez GLL, de Carvalho LM, Hilgemann M, Brainina KZ, Kahlert H, Jack RS, Minh DT (2007) Indirect electrochemical sensing of radicals and radical scavengers in biological matrices. Angew Chem Int Ed 46(42):8079
Hilgemann M, Scholz F, Kahlert H, de Carvalho LM, da Rosa MB, Lindequist U, Wurster M, do Nascimento PC, Bohrer D (2010) Electrochemical assay to quantify the hydroxyl radical scavenging activity of medicinal plant extracts. Electroanal 22(4):406
Zhu AW, Liu Y, Rui Q, Tian Y (2011) Selective and sensitive determination of hydroxyl radicals generated from living cells through an electrochemical impedance method. Chem Commun 47(14):4279
Brillas E, Sirés I, Oturan MA (2009) Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem Rev 109(12):6570
Toffel P, Henglein A (1977) Polarogram of the free hydrogen atom and of some simple organic radicals. Discuss Faraday Soc 63:124
Schiffrin DJ (1973) Application of the photo-electrochemical effect to the study of the electrochemical properties of radical: CO −2 · and ·CH3. Discuss Faraday Soc 56:75
Benderskii VA, Krivenko AG, Kurmaz VA, Simbirtseva GV (1988) Reactions of normal-alkyl radicals at the mercury-electrode. Sov Electrochem 24(1):141
Eberson L (1999) Problems and prospects of the concerted dissociative electron transfer mechanism. Acta Chem Scand 53(10):751
Ludvik J, Nygård B (1996) Electrochemistry of aromatic diselenides and ditellurides in aprotic media. Preceding formation of mercury-containing compounds. Electrochim Acta 41(10):1661
Ludvik J, Nygård B (1997) Electrochemistry and metal complex formation of organic diselenides. J Electroanal Chem 423(1–2):1
Kurmaz VA, Gultyai VP (2010) Electrode reactions and electroanalysis of organomercury compounds. Russ Chem Rev 79(4):307
Kurmaz VA, Ershler AB (2006) Acid-catalysed degradation of the organomercury intermediates of pentafluorophenylmercury bromide reduction near a mercury electrode. Mendeleev Commun 16(4):234
Rufman NM, Rotenberg ZA (1980) Special kinetic features of the photo-decomposition of organolead compounds at lead electrode surfaces. Sov Electrochem 16(3):309
Wang LM, Zhang JS (2002) Ab initio study of reaction of dimethyl sulfoxide (DMSO) with OH radical. Chem Phys Lett 356(5–6):490
Bartels DM, Lawler RG, Trifunac AD (1985) Electron T 1 measurements in short-lived free-radicals by dynamic polarization recovery. J Chem Phys 83(6):2686
Opstad CL, Melo TB, Sliwka HR, Partali V (2009) Formation of DMSO and DMF radicals with minute amounts of base. Tetrahedron 65(36):7616
Chaudhri SA, Göbl M, Freyholdt T, Asmus K-D (1984) A method to generate and study (CH3)2S+. radical cations. Reduction of Me2SO by H. atoms in aqueous HClO4 solutions. J Am Chem Soc 106(20):5988
Occhialini D, Kristensen JS, Daasbjerg K, Lund H (1992) Estimation of reduction and standard potentials of some allyl and substituted alkyl radicals. Acta Chem Scand 46(5):474
Lund H, Skov K, Pedersen SU, Lund T, Daasbjerg K (2000) On the determination and use of reduction potentials of short-lived radicals. A review. Collect Czech Chem Commun 65(6):829
Krivenko AG, Kotkin AS, Kurmaz VA (2002) Mechanism of electroreduction of intermediates with and without a proton donor. Electrochim Acta 47(24):3891
Acknowledgements
The authors are grateful to Prof. Alexander G. Krivenko for helpful discussion. This work is supported by the Russian Foundation for Basic Research (project 09-03-00598).
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is dedicated to the 70th birthday of Fritz Pragst.
Rights and permissions
About this article
Cite this article
Kurmaz, V.A., Kotkin, A.S. & Simbirtseva, G.V. Laser photoemission generation and electrochemical study of methyl radicals as secondary products of OH radicals capture by dimethyl sulfoxide molecules. J Solid State Electrochem 15, 2119–2126 (2011). https://doi.org/10.1007/s10008-011-1534-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-011-1534-1