
Abstract
A novel amperometric biosensor was fabricated for glucose sensing based on the precursor of a tailor-designed platinum nanoparticle (nano-Pt) modified polycrystalline gold disk-microelectrode (poly-Au DME). The platinum nanoparticles were electrodeposited onto poly-Au DME modified with a submonolayer (Ausm) of cysteine (nano-Pt/Ausm), and its resulting electrocatalytic activity was evaluated by chronoamperometry. By means of self-assembly technique, cysteamine was grafted on cysteine-modified nano-Pt/Ausm to introduce sulfhydryl groups for immobilization of gold nanoparticles (nano-Au) and adsorption of glucose oxidase (GOD, which acts as an enzyme template) at nano-Au. In order to improve the anti-interference ability, diethylenetriaminepentaacetic acid (DTPA), with negatively charged functional groups, was anchored on the modified microelectrode. This well-prepared biosensor shows remarkable electrocatalytic activity and selectivity towards hydrogen peroxide ejected from enzymatic activities, with a pronounced oxidation current at a low positive potential of 0.4 V (vs. Ag/AgCl). Glucose is chronoamperometrically determined, and the linear range is between 0.1 and 50 μM, with a detection limit of 0.01 μM. The response time is less than 5 s. In addition, it exhibits good reproducibility, strong stability, and less interference from other coexistent electroactive species.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The crucial issues of developing novel and highly efficient amperometric enzymatic biosensors are (1) the effective immobilization of bioactive enzymes, such as glucose oxidase (GOD), in matrices by variously designed techniques, and (2) the highly sensitive and selective detection of the reaction products (e.g., H2O2) released from enzymatically catalyzed reactions during glucose mensuration. Due to their rapid response and intrinsic reaction specificity, enzymatic biosensors have received unprecedented applications in numerous research fields, especially for medical diagnostics [1] and environmental monitoring [2], among others.
In the case of enzymatic biosensors for glucose, the effective immobilization of enzymes in matrices with good biological activities is an essential part of the construction. During recent decades, many conscientious approaches have been taken in immobilizing enzymes onto or into various substrate matrices, including adsorption [3, 4], cross-linking [5], electrochemical copolymerization [6, 7], covalent attachment [8], polymeric film entrapment [9], and magnetic particle immobilization [10]. Among these, adsorption is an important method that can be performed simply in conventional situations. However, the most serious drawback that yet needs to be overcome involves the low adsorption of the enzyme molecules. On the other hand, amperometric biosensor detection of H2O2 derived from enzymatic reaction by electrochemical oxidation of H2O2 is more favored because the reduction of H2O2 is very sensitive to the cleanliness of the Pt electrode surface and the reductive currents easily suffer from the interferences from oxidative species, such as dissolved oxygen [11]. However, since the anodic potential commonly used for electrochemical oxidation of H2O2 is usually +0.6 V or higher [6], there will be some concomitant signals from other oxidizable electroactive species, such as ascorbic acid (AA), uric acid (UA), and acetaminophen (AAP), that coexist with glucose in serum. These other compounds could contribute non-negligible interference, especially in clinical analysis. In order to avoid these interferences as much as possible, some efforts have been made to improve biosensor selectivity via detection of H2O2 at relatively lower potentials [5, 12, 13] or through the incorporation of nonconducting polymers as permselective membranes [4, 8].
Since nanomaterials have become a research hotspot for their temptingly unique electronic, optical, and catalytic properties, electrochemical research into and applications of nanoparticles have attracted much attention in recent years [14–16]. Nanoparticle-modified electrodes usually exhibit more advantages than do macroelectrodes in terms of electroanalytical behaviors, showing higher catalytic performance, improvement of mass transport, etc., which are chiefly attributable to size effects. As reported previously, nanoparticles, e.g., gold or platinum nanoparticles, are effective matrices for the adsorption and immobilization of enzymes, due to their huge specific surface area [5, 17, 18]. More importantly, nanoparticles possess good biocompatibility that can provide enzyme-friendly platforms for keeping their biologic vitalities during determinations [19, 20]. According to some research [21, 22], nanoparticles, especially the platinum nanoparticles, have been proven to efficiently lower the electrochemical oxidation overpotential of H2O2. As a consequence, the response based on electrocatalytic oxidation of H2O2 for a glucose biosensor is very sensitive to the size of nano-Pt modified on the substrate, due to the significant catalytic performances. Consequently, a high value is placed on technologies for electrochemical deposition of microscale or nanostructure metal particles that utilize tailor-made design methods [23–26]. The crystallite size of the nanoparticles can be controlled by various procedures, such as substrates modified with template layers of agarose gels [23] and submonolayers of thiol compounds [24], or the variation of physical and chemical process parameters [25, 26].
In this work, we have extended our methodology by taking account of the advantageous features of gold nanoparticles as effective immobilization matrices and of GOD as a model enzyme for adsorption, especially on the tailor-made design of a platinum nanoparticle-modified precursor. In this case, platinum nanoparticles were electrochemically deposited onto a polycrystalline gold disk-microelectrode modified with a submonolayer (Ausm) of cysteine. The platinum nanoparticles grew selectively onto the bare fraction of the poly-Au DME that was free from coverage of cysteine (namely, the Au(111) domains of the poly-Au), while the other sections (namely, Au(100) and Au(110)) were under the protection of cysteine, as has been demonstrated by several studies [24, 27, 28]. The objective of the presented method was to capture relatively smaller size platinum nanoparticles and modify Au(100)-enriched gold substrate as a novel and efficient electrocatalytic matrix. This would cooperate with gold nanoparticles during the electrochemical oxidation process of H2O2. In addition, diethylenetriaminepentaacetic acid (DTPA), which consists of negatively charged functional groups, was self-assembled on the modified microelectrode to improve the selectivity of the microbiosensor. This well-prepared biosensor displayed excellent results on the determination of glucose by measuring the oxidation signals of H2O2 at a low positive potential with high sensitivity, short response time, and good selectivity.
Experimental
Reagents and apparatus
GOD from Aspergillus niger (EC 1.1.3.4, type VII-S, 250 U/mg) was purchased from Sigma (USA); d-glucose, l-cysteine (Cys, >98%), 2-mercaptoethylamine (MPE, Cysteamine, >98%), N-hydroxysuccinimide (NHS), and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) were purchased from Shanghai Biochemical Reagent (China); and H2PtCl6, HAuCl4, DTPA, and 30% H2O2 were obtained from Guangzhou Chemical Reagent (China). All the other reagents were analytical grade and used as received. All aqueous solutions were prepared with doubly distilled water. The supporting electrolyte was 0.1 M phosphate-buffered solution (PBS) containing 0.2 M KCl, which was prepared with KH2PO4 and Na2HPO4. Gold nanoparticles were prepared according to the literature [29] with a slight modification. During gold nanoparticles synthesis, 2.50 mL of 1.0% trisodium citrate was added to 100 mL of boiling 0.01% HAuCl4 solution.
A CHI 660B electrochemical workstation (Chenhua Instruments, China) was used to perform the electrochemical detection (unless otherwise specified) at room temperature (ca. 25°C). The electrochemical cell was a classical three-electrode system with a gold disk-microelectrode as the working electrode, an Ag/AgCl (saturated with KCl solution) as the reference electrode, and a platinum wire as the auxiliary electrode. Electrochemical impedance spectroscopy (EIS) measurements were carried out by Autolab PGSTAT12 (Eco Chemie, The Netherlands) in PBS (pH 6.8) containing 10 mM K3[Fe(CN)6] and 0.2 M KCl, and the frequency range was 1.0 × 10−3 to 1.0 × 104 Hz.
Preparation of a tailor-designed platinum nanoparticle-modified gold microelectrode
Before modification, the polycrystalline gold disk-microelectrode (25 μm diameter) was polished successively with 0.3 and 0.05 μm α-alumina slurry on chamois and then sonicated in ethanol and water for 5 min. The pretreated microelectrode was then cleaned electrochemically in 0.5 M H2SO4 by cyclic voltammetric (CV) sweeping from −0.2 to 1.5 V at scan rate of 100 mV/s until reproducible CV scans were recorded. A platinum disk-microelectrode (25 μm diameter) was prepared in a similar way to the poly-Au DME by mechanical polishing and electrochemical scanning from −0.2 to 1.3 V at 100 mV/s until the CV characteristic revealing a clean Pt electrode.
The gold microelectrode was immersed in 0.05 M H2SO4 solution containing 1 × 10−3 M H2[PtCl6], and a potential for step electrolysis from 0.5 to 0.1 V was applied for 25 s. Platinum nanoparticles were electrodeposited onto the unmodified (nano-Pt/Au) and modified with a submonolayer (nano-Pt/Ausm) of cysteine poly-Au DME according to the literature [26], with minor modification. The latter was done for the purpose of preferential electrodeposition of nano-Pt onto specific surface domains of the poly-Au DME (i.e., onto the facets of Au(111)) and was accomplished by the following plotted strategy. First, the poly-Au DME was soaked in 0.1 M HCl solution containing 0.1 M cysteine for 2 h, to obtain a full monolayer of cysteine-modified microelectrode [30], and then was thoroughly washed with water to remove the nonchemisorbed cysteine molecules. Secondly, part of the cysteine monolayer was reduced for desorption by sweeping the potential once between −0.3 and −0.8 V in 0.5 M KOH solution. This step was aimed at the removal of weakly chemical bounded cysteine molecules, which had adsorbed at the facets of Au(111) and at the retention of strongly chemical-bonded cysteine molecules that had assembled at the sections of Au(100) and Au(110). This procedure resulted in the successful fabrication of a poly-Au DME surface modified with a submonolayer of cysteine (Ausm) [26–28]. Finally, the platinum nanoparticles were electrodeposited onto this tailor-made Ausm microelectrode to obtain a nano-Pt/Ausm where platinum nanoparticles grew especially onto the bare fraction of the poly-Au electrode (i.e., Au(111) domains). The electrochemical growth of platinum nanoparticles during electrodeposition on the poly-Au DME modified with the cysteine submonolayer is supposed to take place onto (1) the bare Au(111), (2) Au(100) and Au(110), which were below cysteine, and (3) atop the cysteine molecules (i.e., the –NH2 or –COOH terminal groups of cysteine form a metalized self-assembled monolayer (SAM)). Fortunately, in the aforementioned conditions, the platinum nanoparticles are believed to grow selectively onto the bare Au(111) domains [26] because: (1) the possible further growth underneath cysteine could be neglected for the short electrodeposition time (25 s) and (2) the metallization of SAM was more laborious due to the high resistance of cysteine compared with the dominant deposition onto bare Au(111).
The residual cysteine molecules (i.e., adsorbed on Au(100) and Au(110)), as well as the supposedly existent metalized SAM of the prepared nano-Pt/Ausm microelectrode, were completely removed by CV sweeping from −0.2 to 1.5 V in 0.5 M H2SO4 at a scan rate of 100 mV/s for several scans and which was confirmed by the clearly characteristic CV curves obtained in 0.5 M H2SO4.
Construction of the biosensor
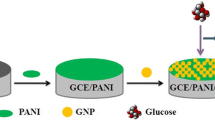
Figure 1 shows the stepwise fabrication process of a glucose biosensor on the nano-Pt/Ausm precursor. The processes for bare Au, bare Pt, and nano-Pt/Au precursor are the same, based on the self-assembly technique. The tailor-made platinum nanoparticle-modified poly-Au DME was immersed in a 0.1 M HCl solution containing 0.1 M cysteine for 2 h to give a cysteine SAM, then washed with water thoroughly to remove the nonchemisorbed cysteine. The cysteine-modified electrode was then dipped into PBS (pH 5.6) in the presence of 15 mM EDC and 30 mM NHS for 1.5 h at room temperature. This step turned the carboxyls of the cysteine molecules into active carbodiimide esters. The activated electrode was subsequently placed into 0.1 M cysteamine PBS (pH 5.6) for 1.5 h, during which the amino-terminal of cysteamine was coupled to the active carbodiimide esters, forming amide bonds. In the meantime, the DTPA solution was activated by the aforementioned EDC/NHS mixture and then linked up with cysteine via the same carboxyl-amine coupling mechanism and washed with water. Thereafter, gold nanoparticles were chemisorbed onto the terminal sulfhydryls (–SH) of this prepared electrode by placing it in a colloidal gold solution for 2 h at 4°C. Finally, GOD was adsorbed onto the surface of the gold nanoparticles by means of soaking the electrode in PBS (pH 5.6) containing 5.0 mg/mL GOD for 2 h at 4°C. The enzyme electrode was washed in gently stirring PBS (pH 5.6) and stored in PBS (pH 5.6) at 4°C until use.

Scheme of the stepwise fabrication process for a glucose biosensor based on the precursor of tailor-designed platinum nanoparticles modified polycrystalline gold microelectrode
Results and discussion
Characterization of different Au microelectrodes
Figure 2 shows the CV responses of bare Au (a), bare Pt (b), nano-Pt/Au (c), and nano-Pt/Ausm (d) microelectrodes in 0.5 M H2SO4 electrolyte. As shown by curve a, there are two characteristic peaks, an oxidation peak extending over a wide potential (ca. 1.05–1.45 V) and a reduction peak occurring at around 0.95 V, corresponding to the solid-state surface redox transition involving the Au/Au oxide [26]. Similarly, curve b shows the characteristic peaks of a Pt/Pt oxide and the adsorption–desorption peaks of hydrogen in the potential range of ca. −0.2 to 0.0 V. In curves c and d, the voltammetric behaviors of nano-Pt/Au and nano-Pt/Ausm microelectrodes are shown, which reveal a combination of the two characteristic peaks of bare Au (curve a) and bare Pt (curve b). This indicates that the Au substrate is partially covered with electrodeposited nano-Pt to a different extent. In the case of nano-Pt/Ausm electrode, the reduction peak intensity of the Pt oxide monolayer is larger than that obtained for the Pt and nano-Pt/Au microelectrode and vice versa, on the reduction peak of the Au oxide monolayer.

CVs of bare Au (a), bare Pt (b), nano-Pt/Au (c), and nano-Pt/Ausm (d) microelectrodes obtained in 0.5 M H2SO4 at 100 mV/s. The microelectrodes are 25 μm in diameter
Another notable feature of the nano-Pt/Ausm electrode is that the adsorption–desorption peaks of hydrogen are much more obvious than for the other electrodes. The differences may be due to different surface coverage of nano-Pt at the electrodes, as well as morphology (i.e., particle size) of the electrodeposited nano-Pt. According to studies reported previously [26], the resulting nano-Pt/Ausm electrode has similar nano-Pt loadings; however, it has larger active surface area than the nano-Pt/Au electrode for smaller size nano-Pt, as demonstrated by the SEM micrographs shown in Fig. 3. The obvious difference in the morphology is the microstructure of electrodeposited platinum particles. The platinum particles (the white spots) electrodeposited onto Ausm (i.e., modified with a submonolayer of thiol) are homogenously distributed in the form of particles and are of relatively smaller size (ca. 70–90 nm, image b) than that electrodeposited on the unmodified Au electrode (size of ca. 100–300 nm in the form of microscale platinum nanoplates, image a). This may explain the larger electrochemically active area obtained at the nano-Pt/Ausm electrode compared to the nano-Pt/Au electrode. Figure 3c shows the transmission electron microscopic (TEM) images of colloidal gold nanoparticle morphology, the average size of gold nanoparticle were 12.25 ± 0.70 nm by counting at 50 particles. Assuming that the size of Au nanoparticles used to load GOD is smaller than that of Pt nanoparticles. X-ray energy dispersive spectrometric (EDS) technique had been used to further characterize in detail the Au and cysteine/nano-Pt/Ausm assembled electrode, and their corresponding elemental profiles were shown in Fig. 4. The element analysis data for cysteine/nano-Pt/Ausm electrode were Au 97.59, Pt 1.23, and S 1.18 wt%, which indicated that Pt nanoparticles had been coated on gold electrode via cysteine.

Top-view SEM images of nano-Pt/Au (a) and nano-Pt/Ausm (b) microelectrodes prepared as mentioned in Fig. 2, and the TEM images of colloidal gold nanoparticle morphology

EDS of Au (a) and cysteine/nano-Pt/Ausm (b) electrode
Amperometric oxidation of H2O2 at nano-Pt-modified Au microelectrodes
H2O2 is a typical reaction product of the GOD-catalyzed oxidation of glucose in the presence of dissolved oxygen, and was examined by chronoamperometry in PBS (pH 6.8, 10 mL). It is apparent from electrooxidation currents of H2O2 in Fig. 5, for bare Au (curve a) and bare Pt (curve b) microelectrodes, very small amperometric response can be observed. When platinum nanoparticles were electrochemically deposited onto the polycrystalline gold microelectrode, without or with a cysteine submonolayer, the amperometric responses increased markedly compared with bare Au and Pt microelectrodes. The nano-Pt/Ausm (curve d) microelectrode exhibited superior catalytic characteristic towards H2O2 compared with the nano-Pt/Au (curve c). The sensitivity for nano-Pt/Ausm extracted from amperometric i–t curve is 3.6 nA/μM, which is substantially higher than the others, for nano-Pt/Au (2.3 nA/μM), bare Pt (0.42 nA/μM) and bare Au (0.14 nA/μM). This is mainly due to the size effect of the platinum nanoparticles because relatively smaller nanoparticles might provide a larger available active surface and facilitate the electron transfer for H2O2 electrooxidation. The response of the nano-Pt/Ausm microelectrode to H2O2 was very fast (within 5 s).

Amperometric responses of H2O2 at bare Au (a), bare Pt (b), nano-Pt/Au (c), and nano-Pt/Ausm (d) microelectrodes for successive addition (10 μL) of 1.0 × 10−4 M (I), 2.0 × 10−4 M (II), and 3.0 × 10−4 M (III) of H2O2 into gently stirring PBS (pH 6.8, 10 mL) held at 0.4 V. Inset CVs obtained on the nano-Pt/Ausm microelectrode in the absence (e) and presence (f) of 2.0 × 10−4 M H2O2 at scan of 50 mV/s
For comparison, CVs obtained on the nano-Pt/Ausm microelectrode in the presence (f) and absence (e) of H2O2 are displayed in the inset of Fig. 5 and a visible oxidation peak is observed on nano-Pt/Ausm precursor around +0.4 V. It is evident that the tailor-designed platinum nanoparticles enhance the electron transfer taking place during H2O2 oxidation.
Film assembling and characterization by EIS
EIS is a powerful tool for studying the interface properties of surface-modified electrodes [31]. The semicircular diameter of the impedance spectrum is approximately equal to the electron-transfer resistance (R et) at the electrode surface, which controls the charge transfer kinetics of the redox probe (i.e., Fe(CN) 3−6 in our paper) at the electrode interface. As shown in Fig. 6, for the electrochemical impedance study, the bare nano-Pt/Ausm shows a small semicircle followed by a straight line (curve a), implying low R et to the redox probe. After the formation of cysteine SAM, a barrier to the interfacial charge transfer is in place, as demonstrated by the increasing semicircle diameter (curve b). This may be due to the electrostatic repulsion effect between the negatively charged Fe(CN) 3−6 and cysteine in PBS (pH 5.6) for its isoelectric point (pI) of 5.02. When cysteamine was attached to cysteine SAM, the semicircle diameter increased once more (curve c); the reason may be that the R et increased with the increasing length of the alkyl chain alkanethiol [32]. When DTPA was linked to the amino groups of cysteine, the semicircle diameter increased again, in agreement with our expected results (curve d). This must be ascribed to the negatively charged DTPA in PBS (pH 5.6) for its five different pKa values of 1.9, 2.9, 4.4, 8.7, and 10.5. After the immobilization of gold nanoparticles assembled on the sulfhydryl layer (curve e), the semicircle diameter decreased suddenly. This may be attributed to gold nanoparticles acting as tiny conduction centers that facilitated the electron transfer of the redox probe. Finally, GOD was strongly covalently tied to the gold nanoparticle surface, owing to the lysine residues (–NH2) of GOD. A significant increase of R et (curve f) was obtained because gold nanoparticles were narrowly covered by GOD, due to its nonconductive properties, as well as carrying a net negative charge at pH 5.6 because of its pI = 4.2 [33]. The EIS experiments confirm the success of the assembly process.

EIS of various electrodes obtained in PBS (pH 6.8) containing 10 mM K3[Fe(CN)6] and 0.2 M KCl in the frequency range from 1.0 × 10−3 Hz to 1.0 × 104 Hz. a nano-Pt/Ausm electrode, b electrode a assembled with cysteine, c electrode b been attached with cysteamine, d DTPA linked to electrode c, e immobilization of gold nanoparticles on the electrode d, f GOD covalently tied to the electrode e
CV responses of the microbiosensor towards glucose
The mechanism of glucose assay in GOD-based biosensors depends on the amperometric detection of H2O2, as mentioned above. CVs were recorded at the biosensors derived from Au, Pt, nano-Pt/Au, and nano-Pt/Ausm precursor microelectrodes following injection of 1.0 × 10−5 M glucose into the PBS (pH 6.8, 10 mL), as shown in Fig. 7. For Au (curve a) and Pt (curve b) based enzyme microelectrodes, there were neither reduction peaks nor oxidation peaks by sweeping from −0.2 to 0.6 V at scan rate of 50 mV/s. For the nano-Pt/Au based enzyme microelectrode (curve c), an indefinite and inconspicuous oxidation peak loomed around 0.4 V accompanied by an increasing oxidation current. However, a well-defined oxidation peak is observed around 0.4 V for the nano-Pt/Ausm (curve d) based enzyme microelectrode, which affords a relatively low and wide potential window (ca. 0.35 to 0.45 V) for amperometric detection of glucose. Another significant feature is the remarkable increase in oxidation current, which is mainly ascribed to the electrocatalytic activity of the tailor-designed platinum nanoparticles, due to their inherent features, such as size effects, and high-surface active sites, among others. For comparison, CVs obtained in the absence of glucose for the nano-Pt/Au (curve e) and nano-Pt/Ausm (curve f) based biosensors are shown in the inset of Fig. 7, which shows an invisible background current. The cathodic current in Fig. 7 is also glucose-dependent, which was generated from the electrocatalytic reduction of H2O2. However, anodic currents were measured for glucose determination due to the reason previously mentioned.
Consequently, an operating potential of +0.4 V was chosen for subsequent experiments on the detection of glucose.

CV responses of biosensors based on different precursor substrates of Au (a), Pt (b), nano-Pt/Au (c), and nano-Pt/Ausm (d) obtained in PBS (pH 6.8) in the presence of 1.0 × 10−5 M glucose sweeping from −0.2 to 0.6 V at scan rate of 50 mV/s. Inset CVs obtained in the absence of glucose for nano-Pt/Au (e) and nano-Pt/Ausm (f) based biosensors
Effects of the pH value and temperature on the biosensor
In order to obtain an efficient response for glucose measurements, the influence of pH on oxidation currents of the proposed biosensor was investigated in a range from pH 5.0 to 9.0 at a constant glucose concentration (5.0 μM). The increase in amperometric current from pH 3.0 to 6.0 corresponded to the increase in the bioactivity of immobilized GOD. The maximum current was observed close to pH ∼ 7. The subsequent decrease in amperometric response at higher pH (8–9) was probably due to the denaturation of GOD. According to the optimum pH value of GOD [34], a neutral pH value of 6.8 was chosen as the working PBS condition.
The effect of temperature on the performance of biosensor was studied in constant glucose concentration (5.0 μM) at the range between 15°C and 60°C. The response currents increased with the increase of temperature and the maximum response current was observed at about 45°C. Considering both lifetime and ease of operation of the biosensor, room temperature (ca. 25°C) was employed as the optimum temperature for our glucose determinations.
Amperometric response and determination of glucose at nano-Pt/Ausm biosensor
Figure 8 shows the amperometric responses of the nano-Pt/Ausm biosensor microelectrode towards glucose at +0.4 V in PBS (pH 6.8, 10 mL) solution following successive additions of different concentrations of glucose under gently magnetic stirring. The biosensor displayed a rapid and pronounced increase in current in response to glucose addition. The time required to reach 95% steady state response was within 5 s, which suggests excellent electrocatalytic characteristics of this microbiosensor.

Amperometric response of glucose at the nano-Pt/Ausm biosensor for successive addition (10 μL) of 1.0 × 10−3 M (a), 2.0 × 10−3 M (b) and 3.0 × 10−3 M (c) glucose into gently stirred PBS (pH 6.8, 10 mL) held at 0.4 V. Inset plot of chronoamperometric oxidation currents vs glucose concentration
The electrochemical measurements of glucose were carried out by means of chronoamperometry. Different aliquots of 1.0 × 10−3 M glucose solution were successively injected into 10.0 mL PBS (pH 6.8) under gentle stirring, and the amperometric i–t curve corresponding to the oxidation currents was recorded at 0.4 V. The plot of oxidation currents vs. glucose concentration is shown in the inset of Fig. 8, fitting a linear relationship over the range of 0.1 to 50 μM, with a correlation coefficient of 0.9987. The biosensor exhibits a sensitivity of 0.29 nA/μM and a lower detection limit of 0.01 μM (S/N = 3). The low detection limit is 1/270 of that for a chitosan/GOD/nano-Au particle biocomposite [6] and 1/670 for multilayer films via layer-by-layer self-assembly of MWCNts, nano-Au, and GOD on a Pt electrode [4]. This clearly demonstrates the high catalytic properties of these tailor-designed platinum nanoparticles and the synergistic effect between platinum and gold nanoparticles.
This high sensitivity for glucose determination indicates that the tailor-designed platinum nanoparticle-modified precursor cooperating with gold nanoparticles provides a synergistically efficient substrate for the oxidation of H2O2 released from enzymatically catalyzed reactions.
Reproducibility, repeatability, and stability
The repeatability and reproducibility of the biosensor were also studied. The relative standard deviation (RSD) of the biosensor repeatability was 4.3%, as estimated from the amperometric current of 5.0 μM glucose for seven successive assays. Reproducibility of the fabrication procedure (the operational stability) under the same conditions was also assessed from different batches and a RSD of 5.6% (n = 7) was obtained for detection of 5.0 μM glucose. The stability of the biosensor stored in PBS (pH 6.8) at 4°C was investigated by periodically recording its current in response to 5.0 μM glucose. The response current was unchanged over a 2-week period, and the electrode retained 93% of its initial sensitivity after 1 month. Good storage stability was mainly due to the natural features of gold nanoparticles for GOD molecule immobilization, characterized by low loss of activity or amount.
Interference test and sample analysis
The common electroactive interfering substances that coexist with glucose in physiological samples (e.g., blood serum) include AA and UA, and usually cause problems in the accurate determination of glucose. Consequently, the selectivity of the proposed the biosensor was tested. Amperometric i–t curves were performed to record the response signals of AA, UA, and AAP during the determination of glucose in the absence or presence of the interference-suppressing agent, DTPA. As shown in Fig. 9, injection of 10 μL 1.0 × 10−3 M glucose into PBS (pH 6.8, 10 mL) caused an immediate increase for the oxidation current at both biosensors. However, injection of 10 μL 1.0 × 10−3 M AA, UA, or AAP did not cause any obvious interference on response current at the biosensor modified with DTPA (curve a) compared with the one modified without DTPA (curve b). The responses to glucose for both biosensors were very similar, indicating that the DTPA modifier did not create steric hindrance or diminish the oxidation current. The selectivity ratio within the linear regime of sensor operation was 1:20, 1:30, and 1:30 for AA, UA, and AAP, respectively.

Amperometric responses of glucose, AA, UA, and AAP on the biosensor modified with (curve a) or without (curve b) DTPA in PBS (pH 6.8, 10 mL) held at +0.4 V
DTPA contains a high concentration of negatively charged functional groups (i.e., –COO) in PBS (pH 6.8); therefore, the negatively electrostatic interaction between anions (e.g., anionic ascorbate) and DTPA provides a degree of charge-based discrimination for the produced H2O2 over anions. Good selectivity of the biosensor could also partially be attributed to the low working potential of +0.4 V. Employment of an anti-interference agent grafted onto the modified biosensor retained a high selectivity for glucose determination, without the need for introduction of mediators or permselective membranes. This is a distinct advantage of the proposed biosensor that makes it competitive with or better than those reported previously [4, 8, 22].
In order to test the practical applicability of this biosensor for real samples analyses, the glucose in blood serum was determined. Before determination, 20 μL of the serum samples was added with the help of microinjector into the measuring chamber and diluted with PBS (pH 6.8) to obtain a total 5.0 mL volume. This was then analyzed without any further pretreatment. As a control, the GOD method was employed according to a previous protocol [35]. The glucose concentration levels in sera determined by the proposed biosensor were in agreement with the values determined by the spectrophotometry method and the results are shown in Table 1. Analytical recoveries of the glucose added into blood serum samples were from 95.0% to 97.2%. All experimental results demonstrated the promising capability of this novel biosensor in practical application.
Conclusions
A novel and intriguing route for glucose biosensor fabrication was presented. It is based on immobilization of GOD by adsorption of gold nanoparticles on a precursor of tailor-designed platinum nanoparticle-modified polycrystalline gold microelectrode. Highly activated by combining gold nanoparticles with tailor-designed platinum nanoparticles, the resulting glucose biosensor holds a synergistic electrocatalytic effect for the oxidation of hydrogen peroxide, derived from the GOD production during glucose oxidation, at a low positive potential. In addition, the anti-interference ability was improved significantly due to the negatively charged discrimination of DTPA.
References
Vo-Dinh T, Cullum B (2000) Fresenius J Anal Chem 366:540–551
Rodriguez-Mozaz S, de Alda MJ Lopez, Barceló D (2006) Anal Bioanal Chem 386:1025–1041
Yan W, Feng X, Chen X, Hou W, Zhu J (2008) Biosens Bioelectron 23:925–931
Wu B, Hou S, Yin F, Zhao Z, Wang Y, Wang X, Chen Q (2007) Biosens Bioelectron 22:2854–2860
Kang X, Mai Z, Zou X, Cai P, Mo J (2007) Anal Biochem 369:71–79
Luo X, Xu J, Du Y, Chen H (2004) Anal Biochem 334:284–289
Xue MH, Xu Q, Zhou M, Zhu J (2006) Electrochem Commun 8:1468–1474
Manesh KM, Kim HT, Santhosh P, Gopalan AI, Lee KP (2008) Biosens Bioelectron 23:771–779
Cosnier S (1999) Biosens Bioelectron 14:443–456
Li J, Gao H (2008) Electroanalysis 20:881–106
Horrocks BR, Schmidtke D, Heller A, Bard AJ (1993) Anal Chem 65:3605–3614
Yang M, Yang Y, Liu Y, Shen G, Yu R (2006) Biosens Bioelectron 21:1125–1131
Li J, Yu J (2008) Bioelectrochemistry 72:102–106
Wang J (2005) Analyst 130:421–426
Shipway AN, Katz E, Willner I (2000) ChemPhysChem 1:18–52
He L, Toh CS (2006) Anal Chim Acta 556:1–15
Tang H, Chen J, Yao S, Nie L, Deng G, Kuang Y (2004) Anal Biochem 331:89–97
Bahshi L, Frasconi M, Tel-Vered R, Yehezkeli O, Willner I (2008) Anal Chem 80:8253–8259
Xiao Y, Patolsky F, Katz E, Hainfeld JF, Willner I (2003) Science 299:1877–1881
Yehezkeli O, Yan YM, Baravik I, Tel-Vered R, Willner I (2009) Chem Eur J 15:2674–2679
Miscoria SA, Barrera GD, Rivas GA (2002) Electroanalysis 14:981–987
Chu X, Duan D, Shen G, Yu R (2007) Talanta 71:2040–2047
Hasse U, Scholz F (2006) J Solid State Electrochem 10:380–382
Awad MI, El-Deab MS, Ohsaka T (2007) J Electrochem Soc 154:B810–B816
Natter H, Hempelmann R (2003) Electrochim Acta 49:51–61
Liu H, Favier F, Ng K, Zach MP, Penner RM (2001) Electrochim Acta 47:671–677
El-Deab MS, Arihara K, Ohsaka T (2004) J Electrochem Soc 151:E213–E218
El-Deab MS, Ohsaka T (2003) Electrochem Commun 5:214–219
Doron A, Katz E, Willner I (1995) Langmuir 11:1313–1317
El-Deab MS, Ohsaka T (2004) Electrochim Acta 49:2189–2194
Ehret R, Baumann W, Brischwein M, Schwinde A, Stegbauer K, Wolf B (1997) Biosens Bioelectron 12:29–41
Avila A, Gregory BW, Niki K, Cotton TM (2000) J Phys Chem B 104:2759–2766
Swoboda BEP, Massey V (1965) J Biol Chem 240:2209–2215
Weibel MK, Bright HJ (1971) Biochem J 124:801–807
Huggett ASC, Nixon DA (1957) Biochem J 6:12–19
Acknowledgements
The authors gratefully acknowledge the financial support received from the National Nature Science Foundation of China (20665003) and the Nature Science Foundation of Guangxi Province (No. 0728214).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, J., Yu, J., Wei, X. et al. A sensitive and selective biosensor activated by tailor-designed platinum nanoparticles electrodeposited onto a gold microelectrode. J Solid State Electrochem 15, 1129–1137 (2011). https://doi.org/10.1007/s10008-010-1178-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-010-1178-6