
Abstract
In the present work, the aluminum electrode surface was modified with a thin layer of palladium by dipping its polished surface in a 25% ammonia solution of PdCl2. The electrocatalytic ability of the modified electrode towards the paracetamol (PCT), ascorbic acid (AA), and codeine (CO) oxidation was investigated using cyclic voltammetry, chronoamperometry. The number of electrons for rate-determining step and diffusion coefficient of PCT, AA, and CO were determined. The electro-oxidation pathway and kinetics, including transfer coefficient α and diffusion coefficient D, are estimated. The results indicated that the simultaneous voltammetry of the three compounds was possible in a mixture solution.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In electroanalysis, the choice of an electrode substrate is greatly influenced by the morphological aspect of its surface because the electrochemical activity strictly depends on the actual chemical and physical status of the electrode surface. In fact, on conventional bulk electrode materials, there are still some problems such the slow electron transfer reaction and electrode poisoning effect. In order to improve the catalytic performance and to decrease the poisoning effect of electrode, the use of appropriately designed chemically modified electrode was considered. The feasibility of deposing metallic particles into organic polymer provides a good physical dispersion of the catalytic centers leading to a highly active electrode surface [1–9]. It is also known that the physical and chemical properties of a metal thin layer on a foreign substrate are different from that of bulk metal [10, 11]. In comparison with other noble metals, palladium displays a higher electrocatalytic activity towards some organic compounds in acidic and alkaline electrolytes through its capability for hydrogen adsorption and dissolution [12–20]. In this regard, the electrocatalytic characteristics of fine palladium particles supported on glassy carbon, carbon fiber, or carbon paste electrodes have been examined towards the hydrogen evolution [20, 21], hydrogen peroxide reduction [22], hydroxyl amine and formaldehyde [23] and oxalic acid oxidation [24]. The electro-oxidation of ascorbic acid on the palladium-coated ore electrode was also reported [25, 26]. Recently, we have reported the preparation and utilization of the aluminum electrode coated by the fine metallic palladium particles for synthesis of some organic and inorganic modifier on the Al electrode surface [27–29]. The palladium-coated Al electrode modified by Prussian blue was also used for the electrocatalytic oxidation and voltammetric or amperometric determination of some analytes [30–32]. The present work deals with the use of an aluminum substrate, modified its surface by metallic palladium particles, designed as Pd/Al electrode for electrocatalytic oxidation and subsequent simultaneous voltammetry of paracetamol (PCT), ascorbic acid (AA), and codeine (CO).
Experimental
Chemicals
The commercial aluminum bar with purity of 99.9 ± 0.1% was used as substrate for electrode matrix. Palladium chloride, ammonia, acetic acid, sodium hydroxide, ascorbic acid, codeine, and paracetamol of analytical grade were used as received.
Instrumentation
All voltammetry or chronoamperometry experiments were performed using a potentiostat/galvanostat AUTOLAB, model PGSTA30. The working electrode was a homemade aluminum disk electrode prepared from an aluminum bar with a purity of 99.9 ± 0.2%. The aluminum bar was mounted in a Teflon tube, exposing a geometrical area of 0.071 cm2, which was modified as detailed below. A conventional three-electrode cell, consisting of a Pd/Al-modified electrode as the working electrode, saturated calomel electrode as the reference electrode and a platinum wire as the counter electrode, were used at room temperature. Scanning electron microscopy (SEM) was performed on a LEO 440i Oxford instrument.
Electrode preparation
The aluminum surface fitted in a Teflon tube [33] was polished, first by a medium emery paper (320-grit) and then by a fine grade (1,500-grit) to expose a relatively mirror surface. The polished surface is cleaned by dipping in 1 M HNO3 for about 1 min and then rinsed with doubly distilled water. The electroless deposition of metallic palladium was carried out by dipping the cleaned surface in 2 mM PdCl2 dissolved in 25% ammonia solution with forming Pd(NH3)2Cl2 complex (plating solution) for 4 min [27]. In order to improve electrochemical behavior of Pd/Al-modified electrode, the electro-pretreatment of the freshly prepared electrode by cycling its potential between 0 and 1.3 V with scan rate of 100 mV s−1 for 60 cycles.
Result and discussion
Characterization of the modified electrode
Figure 1 shows the surface morphology (i.e., SEM) of the Al and Pd/Al electrodes. Image A shows the structure of the bare aluminum surface immediately after polishing with emery paper grade 1,500. As seen in image A, the surface is almost smooth and uniform without any holes or cavities. Image B shows the surface of the same electrode after pretreatment in PdCl2 solution and then conditioning by cycling its potential between 0.0–1.3 V for 60 cycles. As seen in image B, the Pd crystals formed on the aluminum surface, which are settled almost uniformly on it, and they expose a rather developed surface area. Figure 2 shows the cyclic voltammograms of the Pd/Al electrode in 0.25 M KNO3 of various pHs (adjusted by sulfuric acid or acetic acid or sodium acetate). As seen in Fig. 2, at positive scan of potential, no sharp and well-defined anodic peak was observed, whereas during the reverse scanning of potential a stripping voltammogram appeared. This may be attributed to metallic palladium oxidation in anodic cycling and subsequent formation of different variety of palladium oxides such as PdO, PdOH, Pd(OH)2, PdO2, and PdO3 [16] on the electrode, which is stripped during the reverse potential scan for re-formation of a clean surface of palladium. Note that the relevant amounts of these oxides are dependent on the polarization time, applied potential, and especially solution pH. A plot of \( E_{\rm{p}}^{\rm{c}} \) versus pH shown in inset of Fig. 2 is linear with a negative and non-Nernstian slope of 0.082, indicating the simultaneous stripping of different palladium oxides from the electrode surface. This investigation led us to select an electrolyte solution of neutral pHs (e.g., pH 6) where the electrode is bare in potential range between −0.1 and 1 V.

SEM images of a Al electrode surface immediately after polishing. b Pd/Al after conditioning by 60 cycles potential scanning between 0.0–1.3 V

Cyclic voltammograms of Pd/Al-modified electrode at various pHs of 1–8 in 0.25 M NaNO3 + 0.5 CH3COO- solutions of various pHs, (inset) plot of E p versus pH, scan rate: 50 mV s-1
Electrochemical behavior of PCT
The cyclic voltammograms of the Pd/Al-modified electrode recorded at potential scanning range 0 to 1 V in the absence and presence of PCT in 0.5 M KNO3 + 1 M acetate buffer of pH 6 are shown in Fig. 3. As seen in Fig. 3, in the absence of PCT, no peak was observed (curve a). Upon the addition of 3 mM PCT, an anodic peak at 0.67 V appeared (curves b). The peak current increased with scan rate (not shown), and the plot of I p versus square root of the scan rate v 1/2 is linear of regression equation \( {I_{\rm{p}}} = \left( {\hbox{mA}} \right) = 0.051 + 0.054{v^{1/2}}\left( {{\hbox{mV}}{{\hbox{s}}^{ - 1}}} \right) \), suggesting that the reaction is mass transfer-controlled. The peak potential for the oxidation of PCT shifts to more positive potentials with increasing the scan rate as expected for a totally irreversible diffusion-controlled redox process. On the other hand, the peak current increased with increasing PCT concentration, and the calibration curve is linear in the concentration range 0.29–3.3 mM, with a correlation coefficient of 0.9998.

Typical cyclic voltammograms of Pd/Al-modified electrode in 0.25 M NaNO3 + 0.5 CH3COO- solutions of pH 6 in the absence (a) and in the presence (b) of 3 mM PCT, scan rate: 100 mV s-1
The effect of the electrolyte solution pH on the oxidation of PCT at the Al/Pt electrode was investigated. The anodic peak shifted toward more positive potentials with increasing the solution pH, while its height remained almost unchanged. The plot of E p versus pH was linear with slope of 0.034 V between pH 1 and 6. Whereas at pH higher than 6, the E p is maintained independent of the pH.
In order to get information on the final product of the PCT oxidation on the Pd/Al electrode at cyclic voltammetry time scale, we have evaluated the number of electrons (n) involved in the thorough oxidation process. For a totally irreversible diffusion-controlled electrode process, the following equation can be used for evaluation of n [34]:
where n a, α, and A are the number of electrons involved in the rate-determining step, transfer coefficient, and surface area of the electrode, respectively.
According to Eq. 1, the plot of I p versus v 1/2 should be linear with slope of \( 2.99 \times {10^5}n{\left[ {\left( {1 - \alpha } \right){n_{\rm{a}}}} \right]^{1/2}}AC{D^{1/2}} \) allowing calculation of n. For this, the values of (1 – α) n a and D must already be determined. For evaluation of (1 – α) n a, the Tafel plot was drawn using the rising part of the current–voltage curve of 10 mM of PCT monitored at slow scan rate of 10 mV s −1 (Fig. 4). The slope of the Tafel plot was 16.94, yielding a value for (1 – α) n a of 1. Also, the chronoamperometry was used to evaluate the diffusion coefficient of PCT. The current-time profiles were obtained by setting the working electrode potential at 0.8 V in the absence and in the presence of 2, 4, and 6 mM PCT (not shown). At long experimental times (t > 1.5 s or t −1/2 < 0.5), where the electro-catalyzed oxidation rate of PCT exceeds that of its diffusion, the current has a diffusional nature. In this region, the plot of I versus t −1/2 gives a straight line, and slope of such a line can be used for the estimation of the diffusion coefficient D of PCT. From the slope of this plot, the value of D found was 6.1 × 10−6 cm2 s−1. Based on Eq. 1, the plot of I p versus v 1/2 is linear with slope of 0.064. Using such a slope value and considering (1 – α) n a = 1, D = 6.1 × 10-6 cm2 s−1, A = 0.125 cm2, and C = 1 × 10−5 mol /cm3, it is found that the number of electrons involved in thorough anodic oxidation of PCT is 2.17 (about 2 per mole).

Tafel plot obtained for rising part of the cyclic voltammograms recorded for 10 mM PCT at pH 6 with scan rate of 10 mV s-1 supporting electrolyte as in Fig. 3
On the basis of the results obtained from cyclic voltammetry, we can conclude that the oxidation of PCT on the Pd/Al electrode is a two-electron one-proton irreversible process as follows:

Electrochemical behavior of AA
In order to evaluate the electrocatalytic activity of the Pd/Al-modified electrode towards the ascorbic acid electro-oxidation, the cyclic voltammograms were recorded in the absence and presence of ascorbic acid in 0.5 M KNO3 solutions of pH 6 adjusted by 1 M acetic/acetate solution at potential scanning range 0 to 1 V and are shown in Fig. 5. As seen in Fig. 5 in the absence of ascorbic acid, not any peak was discernible (curve a). Upon the addition of 10 mM ascorbic acid, only an anodic peak at 0.2 V appeared (curves b). The plot of peak current against square root of the scan rate showed a good linear relationship of slope 0.038 and correlation coefficient better than 0.999; so, it can be stated that the electro-oxidation of ascorbic acid at the Pd/Al-modified electrode is diffusion-controlled. The calibration graph for ascorbic acid in the concentration range 1 to 100 m M was linear with a correlation coefficient of 0.999 (not shown). The effect of the electrolyte pH on the electro-oxidation of ascorbic acid was investigated by cyclic voltammetry of 10 mM ascorbic acid in 0.5 Na2SO4 + 1 M acetic/acetate. The pH was changed between 1.0 and 8, using a concentrated HNO3 or NaOH solution. Figure 6a shows the relevant results. As it can be seen in Fig. 6, the anodic peak is shifted towards more positive potentials with decreasing the solution pH. The plot E p versus pH (Fig. 6b) showed two linear segments with two Nernstian slopes of 59 (pH < 4) corresponding to a two-electron two-proton process and 28 (4 < pH < 8) relating to a two-electron one-proton oxidation.

Typical cyclic voltammograms of Pd/Al-modified electrode in 0.25 M NaNO3 + 0.5 CH3COO- solutions of pH 6 in the absence (a) and in the presence (b) of 10 mM ascorbic acid, scan rate: 100 mV s-1

a Cyclic voltammograms of 10 mM AA solution on the Pd/Al-modified electrode at various solution pHs of 1–8. b Plot of Ep versus pH, supporting electrolyte and scan rate as in Fig. 5
For getting information on the rate-determining step, a Tafel plot was drawn, derived from data related to the rising part of the current–voltage curve at scan rate of 10 mV s−1 for 10 mM of ascorbic acid (Fig. 7). A slope (b = 2.3RT/(1 − α) n a F) of 116 mV/decade was obtained, indicating a one-electron transfer to be rate-limiting step assuming a transfer coefficient of α = 0.5. The total number of electrons for whole oxidation process of ascorbic acid was about 2 that was determined from slope of I p versus v 1/2 (0.038) according to relation 1 considering \( \left( {1 - \alpha } \right){n_\alpha } = 0.5,D = 4 \times {10^{ - 6}}{\hbox{c}}{{\hbox{m}}^2}\;{{\hbox{s}}^{ - 1}} \) determined by chronoamperometry at 0.4 V in ascorbic acid solution of pH 6.

Tafel plot obtained for rising part of the cyclic voltammogram recorded for 10 mM AA at pH 6 with scan rate of 10 mV s-1, supporting electrolyte as in Fig. 3
On the basis of results obtained from above study, the oxidation pathway of ascorbic acid at various pHs may be shown as follows:
At 4 < pH < 8
A− shows anion radical of H2A. Note that the formation of A− during anodic oxidation of ascorbic acid was also confirmed by EPR spectroscopy [35].
Electrochemical behavior of CO
In order to evaluate the electrocatalytic ability of the Pd/Al-modified electrode towards the CO oxidation, the cyclic voltammograms were recorded in the absence and presence of CO in 0.25 M KNO3 of pH 4 and presented in Fig. 8. As seen in Fig. 8, in the absence of CO, not any peak was observed (curve a). Upon the addition of CO, an anodic peak at 0.95 V appeared (curves b). The height of this peak increased with CO concentration. The calibration graph of codeine is linear in the concentration range 3 to 40 mM, with a correlation coefficient of 0.9998. The plot of the cyclic voltammograms peak current I p versus v 1/2 should be linear, and the slope allows determining n. For this evaluation, the values of (1 – α) n a and D must already be determined. For evaluation of (1 – α) n a, the Tafel plot was drawn using the rising part of the current–voltage curve of 10 mM of CO monitored at slow scan rate of 10 mV s−1. The slope of the Tafel plot was 16.94, yielding a value for (1 – α) n a of 0.3.

Cyclic voltammograms of Pd/Al-modified electrode in 0.25 M NaNO3 + 0.5 CH3COO- solutions of pH 6 in the absence (a) and in the presence (b) of 3 mM CO, scan rate: 50 mV s-1
Also, the chronoamperometry as an effective electrochemical technique was used to evaluate the diffusion coefficient of CO. The current-time profiles was recorded by setting the working electrode potential at 1.2 V at Pd–Al electrode in the absence (curve a) and in the presence of 5 mM CO (note shown). At long experimental times (t > 1.5 s or t −1/2 < 0.5), where the electro-catalyzed oxidation rate of CO exceeds that of CO diffusion, the current has a diffusional nature. In this region, the plot of I versus t −1/2 gives a straight line, and slope of such a line can be used for the estimation of the diffusion coefficient D of the substrate. From the slopes of this plot, the value of D was found to be 2.9 × 10−5 cm2 s−1.
Based on Eq. 1, the plot of I p of voltammograms versus v 1/2 is linear with slope of 0.358, so it can be stated that the oxidation of codeine on the Pd/Al-modified electrode is diffusion-controlled. Using such a slope value and considering (1 – α) n a = 0.3, D = 2.9 × 10−5 cm2 s−1, A = 0.071 cm2, and C = 5 × 10−6 mol/cm3, by assuming a = 0.7, it is found that the number of electrons involved in the thorough anodic oxidation of CO is about 2 per 2 moles of CO, confirming that, bis-CO is the unique oxidation product of CO. These findings lead us to suggest the following simple pathway for the electro-oxidation of CO on Pd–Al electrode:
Or

Simultaneous voltammetry of AA, PCT, and CO
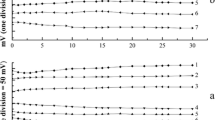
On the basis of the cyclic voltammograms recorded individually for PCT, AA, and CO at pH 1–8, we have found that the peak potential of both compounds shifted to less positive potential as the solution pH increase with same slopes and relevant peaks are of sufficiently different whole pH range, allowing well the separation of the corresponding voltammograms. This study leads us to select a solution pH of 4 as the convenient value for simultaneous voltammetry of PCT, AA, and CO. The measurement is performed in 0.5 M KNO3 + 0.2 M acetate buffer (pH 5). Figure 9 shows the cyclic voltammograms of a solution having the mixture of AA and PCT or PCT and CO of same concentrations at Pd/Al electrode. As can be seen in this figure, three well-defined oxidation peaks of AA, PCT, and CO at about 0.2, 0.6, and 0.95 V, respectively, observed. The peak potential separation of more than 300 mV permits the simultaneous voltammetric measurement of three components of the solution. This is an important role that the Pd/Al electrode plays.

Typical simultaneous cyclic voltammograms of a AA + PCT, b PCT + CO. Peaks: (I) AA, (II) PCT, and (III) CO, supporting electrolyte and scan rate as in Fig. 8
Analytical approach
Figure 9 shows the cyclic voltammograms of the solutions containing the mixture of AA and PCT or PCT and CO at Pd/Al electrode. As can be seen in Fig. 9, two well-defined oxidation peaks of AA, PCT, and CO at about 0.2, 0.6, and 0.95 V observed, respectively. The results show that the peak potential separation is more than 200 mV and the peak current of the three compounds in the presence of fixed concentration of each other is linearly dependant on their concentrations (not shown). This permits the simultaneous cyclic voltammetric determination of AA, PCT, and CO. The performance of the present method was investigated by analysis of two real pharmaceutical preparations (effervescent tablet containing vitamin C and acetaminophen or acetaminophen tablet, containing simultaneously acetaminophen and codeine from local sources). For analysis of these samples, an accurately weighed portion of finely powdered sample obtained from three tablets, equivalent to about 75 mg of drugs was transferred to a 25-ml flask and dissolved with doubly distilled water. A 0.1-ml portion of the solution was diluted with 10 ml of 0.25 M KNO3 electrolyte solution buffered with 0.5 M acetic/acetate at pH 6 in a voltammetric cell and subjected for cyclic voltammetric measurement. The amount of each drug was evaluated by standard addition method. The results of three replicate determinations obtained by the standard addition method confirm that the drug contents found in these samples (0.206 and 0.344 mg per tablet for AA and PCT or 0.485 and 7 mg/tablet for PCT and CO) are consistent with the nominal amounts (200, 330 mg/tablet and 500, 7 mg/tablet ) or determined values by official methods [36, 37] (0.215, 0.402 mg/tablet and 482, 6.85 mg/tablet), respectively. This was confirmed by the statistical analysis, based on Student's t test.
Conclusion
Palladium particles dispersed on an aluminum substrate can be used for the anodic oxidation of PCT, AA, and CO at relatively less positive potentials compared with bulk Pd, Pt, Au, and GC electrodes. The electro-oxidation pathway for the both analytes is similar and the rate-determining step is a one-electron transfer process. The results indicated that the simultaneous voltammetry of the three compounds is possible in a mixture solution. Furthermore, the modified electrode is stable, and its catalytic activities towards electro-oxidation of PCT, AA, and CO reproducible. These make the electrode usable in simultaneous electroanalysis of the both three analytes in real samples.
References
Yet JH, Fedkiw PS (1996) Electrochim Acta 41:221
Wang J, Golden T, Li Y, Agnes L (1992) J Electroanal Chem 333:65
Casella LG, Cataldi TR, Salvi AM, Destimoni E (1993) Anal Chem 65:3143
Laborde H, Leger J-M, Lamy C (1994) J Appl Electrochem 24:219
Wang J, Lu F, Angnes L, Liu J, Sakslund H, Chen Q, Pedrero M, Chen L, Hammerich O (1995) Anal Chim Acta 305:3
Watanabe M, Motoo S (1975) J Electroanal Chem 60:267
Watanabe M, Motoo S (1975) J Electroanal Chem 60:275
Casella IG, Zambonin CG, Perete F (1999) J Chromatogr A 833:75
Yang H, Lu T, Xue K, Sun S, Lu G, Chen S (1997) J Electrochem Soc 144:2302
Henry CR (1998) Sur Sci Rep 31:235
Andonoglou Ph P, Jannakodakis AD (1997) Electrochim Acta 42:1905
Pavese AG, Solis VM, Giordano MC (1987) Electrochim Acta 32:1213
Pavese AG, Solis VM (1991) J Electroanal Chem 301:117
Adzic RR, Sposojevic MD, Despic AR (1978) J Electroanal Chem 92:31
Manzanares MI, Paves AG, Soli VM (1991) J Electroanal Chem 10:159
Becerik I, Kadirgan F (1992) Electrochim Acta 37:2651
Liorca MJ, Feliu JM, Aldaz A, Clavilier J (1994) J Electroanal Chem 376:151
Lu G, Crown A, Wickoviski A (1999) J Phys Chem 103:9700
Rodriguz JA (1996) Sur Sci Rep 24:223
Lubert KH, Guttmann M, Beyer L (1996) Electroanalysis 8:320
Cai X, Kalcher K, Kolbl G, Neuhold C, Diewald W, Ogoreve B (1995) Electroanalysis 7:340
Cai X, Kalcher K, Lintschinger J, Neuhold C, Tykarski J, Ogoreve B (1995) Electroanalysis 7:556
Zhang XG, Murakami Y, Yahilkozawa K, Takasu Y (1997) Electrochim Acta 42:223
Casella IG (1999) Electrochim Acta 44:3353
Matos RC, Augelli MA, Pedrotti JJ, Lago CL, Angnes L (1998) Electroanalysis 10:887
Matos RC, Augelli MA, Lago CL, Angnes L (2000) Anal Chim Acta 404:151
Pournaghi-Azar MH, Dastangoo H (2004) J Electroanal Chem 523:26
Pournaghi-Azar MH, Habibi Biuck A (2007) J Solid State Electrochem 11:505
Pournaghi-Azar MH, Habibi B (2007) J Electroanal Chem 601:53
Pournaghi-Azar MH, Dastangoo H, Ziaei M (2007) J Solid State Electrochem 11:1221
Pournaghi-Azar MH, Ahoor F (2008) J Electroanal Chem 622:22
Pournaghi-Azar MH, Saadatirad A (2008) J Electroanal Chem 624:293
Pournaghi-Azar MH, Razmi-Nerbin H (1998) J Electroanal Chem 456:83
Bard AJ, Faulkner IR (2001) Electrochemical methods, fundamentals and applications, 2nd edn. John Willy, New York
Aldaz A, Aquie AM (1973) J Electroanal Chem 48:532
US Pharmacopoeia XIX (1980), Mack Co, Easton, PA
British Pharmacopoeia II (1998), British Pharmacological Society, Stationary Office Books, London
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pournaghi-Azar, M.H., Kheradmandi, S. & Saadatirad, A. Simultaneous voltammetry of paracetamol, ascorbic acid, and codeine on a palladium-plated aluminum electrode: oxidation pathway and kinetics. J Solid State Electrochem 14, 1689–1695 (2010). https://doi.org/10.1007/s10008-010-1011-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-010-1011-2