
Abstract
Ni–W alloys were electrodeposited onto copper foil from citrate solution. Coatings containing from 11 to 21 at.% W and having 7–52 μm in thickness were obtained. The structure of these alloys was analyzed by X-ray diffraction and by using electron and light microscopy techniques. Alloys with 11 and 15% W are composed of two phases: solid solution of W in fcc Ni and solid solution of Ni in bcc W. An increase in W content in the Ni–W alloys to ca. 18–19% of W resulted in the grain refinement and the transition to amorphous structure. The corrosion behavior of obtained Ni–W and unalloyed Ni coatings was studied in 0.5 M NaCl solution by means of electrochemical impedance spectroscopy, potentiodynamic polarization and light microscopy. Comparing to pure Ni, the obtained Ni–W coatings exhibited a clearly decreased corrosion resistance (in terms of corrosion current density and polarization or charge transfer resistance at the open circuit potential). Despite of the quite wide range of composition of the alloys under test, the related grain refinement, and the transition to the amorphous structure, no clear relation between the corrosion rate and W content was detected. This behavior can be a result of the interplay of the activating effect of grain refinement or preferential dissolution of W from one side and diffusion barrier action or inhibition provided by the surface film of W oxidation products from the other side. The differences observed in the corrosion resistance of Ni–W coatings are more related to their morphological imperfections arising from various deposition conditions than to the W content. Some samples showed a rather non-uniform nature of corrosion (pronounced attack along cracks). An inversion in the dissolution behavior of Ni–W and unalloyed Ni was observed with increasing anodic potential. Contrary to pure Ni, Ni–W coatings were resistant to pitting corrosion in NaCl solution.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ni–W alloys show an unique combination of mechanical (high tensile strength and hardness), tribological (wear resistance), thermal (high melting point, hot strength, oxidation resistance), electrocatalytic (hydrogen evolution), magnetic, and electrical properties [1, 2]. The formation of specific interface between these alloys and the substrate increases their adhesion to plated surfaces [3]. However, the durability under the service conditions is a crucial factor for possible engineering applications of these materials. The excellent functional properties would be useless without the desired corrosion resistance of the Ni–W alloys. Thus, the corrosion properties of Ni–W coatings are of great interest. The passivating ability of W and the transition to nanocrystalline or amorphous structure observed with growing W content should constitute clear prerequisites for a good corrosion resistance of these materials. Indeed, Ni–W alloys are regarded as highly corrosion-resistant. Surprisingly, available reports on corrosion resistance of Ni–W coatings are rather scarce and the main questions related to this topic are still unanswered. In several papers, it is claimed that the Ni–W coatings are more corrosion-resistant than the pure Ni deposits [4, 5]. However, a contrary finding was also reported [6].
Of importance is also the effect of the composition of the Ni–W alloys on their corrosion behavior. The prevailing opinion is that the corrosion resistance of Ni–W coatings increases with W content [4, 7–9]. This view is substantiated by noting that W accumulates on the alloy surface and suppresses the anodic dissolution [1, 4, 10, 11] and that the transition from crystalline to nanocrystalline/amorphous structure takes place [7, 8]. Indeed, in the often-cited paper of Obradovic et al. [4], a decrease in the corrosion current density with increasing W content from 3 to 19 at.% was documented. However, these results were taken under non-stationary conditions, immediately after immersion of the alloy samples into the sulfuric acid solution. The same work reports that when the corrosion test was performed after a longer exposure, the minimum of the corrosion current density was found for the 10%-W coating and was related to its single-phase structure [4]. A maximum corrosion resistance was also observed in NaCl solutions, however, for an intermediate W content, namely ca. 7% [1]. However, in the same work, the corrosion current was found to be almost independent on the composition of the Ni–W coatings deposited at a higher temperature. This behavior was explained by the interplay of the inhibiting chemical action of growing W content and the activating effect of the corresponding grain refinement and the transition to nanocrystalline/amorphous structure (growing density of disordered, intercrystalline regions) [1, 12].
Another open question is the passivation ability of the Ni–W alloys. The experiments performed in sulfuric acid solutions led to opposite conclusions. A remarkable drop in passive current density with an addition of W to Ni was found in those solutions [13]. Sriraman et al. observed an improved passivity of W-rich alloys in 0.5 M H2SO4 [1]. However, Obradovic et al. reported that an increase in W content resulted in a less effective passivation (a sharp increase of current density in the passive state) in the same solution [4].
These conflicting conclusions can be only partly related to conceivable differences in the measurement procedures. The extrapolation of the Tafel segments was mainly used for the determination of the corrosion current density. This method can be highly inaccurate and often yields unreliable results. However, these differences arise, first of all, from the well-known fact that the Ni–W coatings of similar composition can exhibit quite different structure and surface morphology [2, 4, 14]. The corrosion behavior is highly sensitive to these structural features and they, in turn, depend on the electrodeposition conditions (substrate preparation, cathodic current density, bath formulation—especially organic additives surfactants used, temperature, etc.).
Ni–W alloys formed by electrodeposition can contain up to 67% of W [15]. Their structure was not explained satisfactorily yet. The low-W alloys are usually ascribed to a solid solution of W in fcc Ni [1, 14, 16–20]. The intermetallic phase, Ni4W, was another object detected in the Ni–W deposits of similar composition, however, those deposits were plated at an elevated temperature [2, 4, 21]. The non-equilibrium phases, for example supersaturated solid solutions, are also generated by electrodeposition [15, 20, 22, 23]. W-enriched segregations in Ni–W alloys were anticipated in [22, 24]. Thus Ni–W alloys can be composed of a single phase or of several phases [4, 15, 22, 24]. A higher corrosion resistance was reported for the alloys consisting of just one phase [4, 15].
Increasing W content results in the grain refinement and the transition from microcrystalline to nanocrystalline/amorphous structure. The amorphous structure is usually ascribed for the alloys with W content exceeding 20 [25]–22% W [26]. This borderline depends significantly on which surfactants were used [16, 17] and which method of structural investigation was employed. For instance, the crystalline structure was found for relatively W-rich alloy, namely Ni–23W [19]. A multiphase system was observed close to the borderline composition: it consisted of nanocrystallites suspended in the amorphous matrix [24]. The effects of the grain refinement on the corrosion resistance of Ni–W coatings were reported to be either beneficial [7–9] or detrimental [1].
Morphology of the Ni–W alloys affects their corrosion resistance. These coatings show a tendency to cracking. A net of cracks observed on the surface of as-received deposits can result from hydrogen embrittlement [15] or residual stress [27]. Subsequently, the corrosion attack develops mainly along the cracks. Galikova revealed a lowest corrosion rate in 5% NaCl for a less cracked Ni–W coating with 8% W [15]. The deposits with larger W content showed reduced corrosion resistance due to enhanced cracking. There is also a report revealing that the extent of cracking increases with W content only until 20% and it decreases for the alloys with higher than 20% W content [14].
An additional problem, which can affect the reported data on the corrosion of Ni–W, is the possible non-uniformity in the composition of the obtained coatings. Under unfavorable circumstances, local differences in W content can approach 10% [28].
The aim of this work was to examine the corrosion behavior of electrodeposited Ni–W coatings in NaCl solutions and to relate it to the alloy composition and structural state. A wide collection of Ni–W samples was used to achieve conclusions of higher statistical significance. An Ni electrodeposit was used as the reference sample.
Materials and methods
Each Ni–W alloy was electrodeposited galvanostatically from a citrate-based bath onto a piece of copper foil. The solution (1 dm3) contained 79 g of Na3Cit·2H2O, 19.5 g of NaH2PO4·2H2O, 84.6 g of Na2WO4·2H2O, 10.5 g of H3BO3 and 18 g of NiSO4·(NH4)2SO4·6H2O. Deposition agents, namely 50 mg of 2-butin-1,4-diol and 100 mg of ethoxylated phenol (non-ionic detergent), were also added. Prior to the deposition, pH of the bath was adjusted to 8 by respective additions of citric acid or NaOH. All electrodeposition experiments were done at temperatures in the range of 65–70 °C. By varying the deposition current density from 10 to 40 mA/cm2, the coatings containing 11 to 21% of W were obtained. Some modifications in the composition and morphology of Ni–W samples arose also from consecutive deposition experiments (after different charge passed) in the same solutions. The thickness of the coatings was determined by weighing the samples before and after electrodeposition. The EDX data were used in the calculations. The plating time was appropriately adjusted to get the coatings of at least 10 μm in thickness. The reference nickel coatings were deposited from the above bath without addition of tungstate. The parameters of the samples used in subsequent corrosion tests are given in Table 1. The details of copper-substrate preparation and of deposition experiments were given elsewhere [6, 29].
Topography and composition of the samples were examined using a scanning electron microscope, type LEO 435VP (Zeiss, Germany), combined with an EDS detector (Roentec, Germany). The composition of each sample was determined using the EDX method. To assess the possible non-uniformity at the sample surface, the examinations were done at several positions. A scatter in the W content higher than 2% led to the elimination of the sample. The structure of the coatings accepted for the corrosion tests was examined using the XRD method. The X-ray powder diffraction (XRD) patterns were recorded using a Seifert HZG-4 automated diffractometer with the CuKα radiation. The data were collected in the Bragg–Brentano (θ/2θ) horizontal geometry (flat reflection mode) between 30 and 80° (2θ) in 0.04° steps, at the rate of one step per 2 s.
The corrosion resistance of the selected Ni–W coatings was studied in 0.5 M NaCl solution open to air and at 20 ± 2 °C. The solution was prepared from chemical-grade reagents and distilled water. The nickel coatings deposited from the bath without tungstates addition were also tested for comparison purposes. A standard three-electrode cell was employed. A standard calomel electrode (SCE) was used as the reference electrode. A Pt grid of a large area, placed in a separated compartment with a glass frit, was used as the counter electrode. Sectors of the tested coatings of area 0.8 cm2 served as the working electrode. All potentials are expressed versus SCE. Prior to electrochemical corrosion studies the tested coatings were polished with fine emery papers, then rinsed with distilled water, degreased with acetone, and carefully rinsed with methanol.
The corrosion behavior of the Ni–W and Ni coatings was tested by means of chronopotentiometry, electrochemical impedance, and potentiodynamic polarization. An Autolab system (PGSTAT 20 with a FRA module) and the manufacturer’s software were used. The test protocol was as follows. First, before the impedance studies, a chronopotentiometric test was done to monitor the corrosion potential of the immersed samples until its steady state value was reached. The impedance measurement was initiated after 1 h exposure. The impedance data were collected at the corrosion potential, using an excitation potential signal with amplitude of 10 mV, within the frequency range 10 kHz–1 mHz. Ten points were taken per frequency decade. To test the effect of exposure time on the corrosion behavior, the next impedance measurement was repeated after 24 h. Then (after a 27-h immersion), the potentiodynamic polarization curve was registered. The measurement was initiated from the small cathodic polarization (−0.1 V versus E corr) and continued in anodic direction at potential scan of 2 mV/s. The corrosion parameters (corrosion current density and polarization resistance) were derived from the polarization data in the low-polarization region: ±50 mV (the analysis was based on the Butler–Volmer relation). Finally, the corrosion damages of the tested samples were inspected using a light microscope (Nikon Alphaphot-2 YS-2).
The corrosion tests were performed for a number of samples listed in Table 1 for each considered Ni–W composition, and the obtained results were averaged.
Results and discussion
Typical changes in the diffraction patterns of the Ni–W coatings with different W content are shown in Fig. 1. Ni–11W and Ni–15W samples are mainly built of a solid solution of W in fcc Ni which will be called throughout the paper as the Ni–W phase. The diffraction peaks that originated from this phase are shifted to lower values of the 2θ angle, compared to their positions for pure Ni. At the same time, a decrease in the intensities of Ni–W (2 0 0) and Ni–W (2 2 0) peaks with increasing W content is observed, which indicates that both the reduction in the degree of crystallinity and the grain refinement take place. The calculated lattice parameter for the Ni–W phase equals 3.750 Å (for the alloys with 11 and 15% content of W) and is larger than the value of parameter a for pure Ni. This difference conceivably results from a larger atomic radius of W (1.370 Å) which substitutes the host Ni atoms of atomic radius 1.245 Å.

X-ray diffractograms of Ni–W coatings with different tungsten content. Characteristic regions of Ni signals are highlighted
A novelty in the here-reported XRD studies is the appearance of an additional diffraction peak at the 2θ angle equal to ca. 41.5° (d = 2.173 Å) in the Ni–W samples with 11 and 15% of W (see Fig. 1). This finding indicates of the presence of a new phase, not reported for the electroplated materials until now [1, 15, 19, 30], constituted by a solid solution of Ni in W and termed later in the manuscript as the W–Ni phase. This phase is isostructural with metallic bcc W (a = 3.164 Å). The diffraction peak (1 1 0) of the W–Ni phase is located at a larger 2θ angle than the W (1 1 0) peak. This means that the elementary cell is shrunk in this phase (a = 3,073 Å) compared to pure W. A small shoulder, found at a similar position as the above peak (1 1 0) of the W–Ni phase in the XRD spectra of the electrodeposited Ni–20W, was coarsely attributed to the Ni–W alloy [31]. A corresponding, poorly resolved peak has been also shown in the XRD patterns for the alloys with 10.5 and 12.5% W [30], but it was not interpreted by the authors.
Increasing W content in the studied alloys results in a broadening of the diffraction peaks, see Fig. 1, indicating that a larger degree of amorphousness takes place, which is in agreement with the literature data [23, 30]. When the W content exceeds 18%, the peak ascribed to the W–Ni phase as well as the reflections Ni–W (2 0 0) and Ni–W (2 2 0) vanish, and the intensity of the Ni–W (1 1 1) peak decreases. This last reflection is distorted due to overlapping with the peak characteristic for the Cu substrate. The distortion is pronounced for the thinner coatings. Figure 2 presents the relations between the half width and intensity of the Ni–W (1 1 1) peak and W content in the alloy. The half width of this reflection increases and its intensity decreases in the W concentration range from 15–20%. These changes may be attributed to the grain refinement and transition from the microcrystalline to the amorphous state at about 20% W, which is in agreement with [25, 32].

Effect of W content on features of the Ni–W (1 1 1) reflection
The chronopotentiometric data revealed weak dependences of the corrosion potential on time for all the samples, see Fig. 3. The E corr values obtained for Ni–W coatings in the NaCl solution were much more negative than those for pure Ni. A similar dependence is reported in [9]. The corrosion potential decreased gradually with increasing W content in the alloy and the most negative E corr was noted for the sample with the largest W content, namely for Ni–21W. The values of the corrosion potential remained very stable and after 24 h they were practically the same. The E corr results for the Ni–W samples showed a small scatter only. The standard deviations of the averaged E corr values for all alloy samples did not exceed 30 mV.

Time dependence of corrosion potentials of studied samples (numbers at curves denote W content)
Assuming a similar rate of the cathodic reaction (oxygen depolarization) at the tested materials, the more negative E corr of Ni–W samples appears to exclude the beneficial effect of W content in the alloy on the passivity or just the suppression of anodic dissolution of these coatings. The prevailing opinion is that Ni is removed from the Ni–W alloy surface while W remains on it and suppresses its anodic dissolution. However, this finding refers to acidic solutions [1, 4, 10, 11]. In slightly alkaline solutions (e.g., in borate buffer solution of pH = 9.2), Ni–6W exhibits a preferential dissolution of W [33], and conceivably a similar behavior appears in the neutral solution studied in this work.
Slight changes of the corrosion potential with time allowed us to assume that the stability of the corrosion process can be achieved. Therefore, the first impedance measurements were initiated after 1-h immersion. These measurements were also done after 24 h of immersion to assess the effect of exposure time. A comparison of the spectra, typical for each considered Ni–W composition, acquired for both exposure times is shown in the Bode format in Fig. 4.


Impedance spectra (Bode format) for: Ni (a), Ni–11W (b), Ni–15W (c), Ni–18W (d), Ni–20W (e), and Ni–21W (f) after 1- and 24-h exposure times
The impedance response, collected for the Ni coating after 1-h immersion, shows one, very broad and unsymmetric peak of the phase shift with a maximum located between 100 and 101 Hz, see Fig. 4a. Such a shape can be related to overlapping of two peaks corresponding to different phenomena of similar time constants. Indeed, after a longer exposure, the splitting of this peak became evident with an additional impedance phenomenon appearing on the low-frequency side (between 10−2 and 10−1 Hz). The low-frequency impedance values hardly changed with immersion time; so the effect of time on the corrosion resistance of Ni should be considered as minor.
The impedance responses for the Ni–W coatings are significantly different from that for pure Ni and unexpectedly similar to those of all alloy compositions studied. This is illustrated in Fig. 4b–f. The spectra recorded after 1-h exposure exhibit a peak of the phase shift located at the medium-frequency region and an additional response at low frequencies. The medium-frequency peak (MF peak) is well defined and occurs at about 10 Hz for all the Ni–W samples, irrespective of W content. In contrast to this, the low-frequency response (LF response) is very poorly defined. The LF response occurs for the frequencies smaller than 10−2 Hz, which is less than the LF frequency of the shoulder observed for Ni, see Fig. 4a. For the low-W-content alloys, this response looks like a spectrum distortion, see Fig. 4b and d, whereas it is more pronounced for the alloys with higher W content, Fig. 4e and f. The LF impedance values are reasonably similar for all the Ni–W alloys tested.
The impedance spectra acquired after 24 h are distinctly different. The MF peak is shifted to lower frequencies and the LF response becomes more distinguishable. It is clear, that this LF phenomenon is characterized by a very big time constant. After comparing the 24-h results to the data taken after 1 h, it can be said that the LF impedance values do not change much, but the corresponding LF-phase-shift values are larger. Of interest is that all the Ni–W coatings show smaller low-frequency impedance values compared to that of pure Ni sample. This finding suggests that the alloying with W results in a drop in the corrosion resistance.
Evident differences in the shape of the spectra, characteristic frequencies and the impedance values in the impedance data for the Ni–W and Ni coatings indicate that the nature of the corrosion processes of these materials is different. However, in both cases, the acquired spectra contained two time constants. Such impedance responses can be related to a heterogeneous reaction (localized corrosion) on the electrode surface. Indeed, the two time-constant spectra were attributed to non-uniform corrosion on the cracked Ni–W coatings [34]. On the other hand, such impedance spectra can be also interpreted in terms of the charge transfer process accomplished by either reagent adsorption or corrosion-product precipitation or porous-like behavior. The impedance diagrams composed of two capacitive responses with very similar frequency characteristics were also observed for galvanized steel covered with thin films of chromates [35] and molybdates [36].
The recorded impedance data were modeled using an equivalent electrical circuit shown in Fig. 5. It is conceivable that the parallel combination of R 2 and CPE1 is related to the MF peak, whereas the parallel combination of R 3 and CPE2 corresponds to the LF response. However, the physicochemical meaning of these circuit components, except the R 1 component (solution resistance), was unclear a priori and could be clarified after obtaining the fitting results. The obtained values of a given circuit parameter served as a criterion to prescription of this parameter to the specific process.

Electrical equivalent circuit used for analysis of impedance spectra. R 1 solution resistance, R 2 charge transfer resistance, CPE1 constant phase element describing the double-layer capacitance, R 3 and CPE 2 approximate the diffusion impedance through the surface film
The values of the selected parameters, derived from the analysis of the collected impedance spectra, are shown together with the corresponding standard deviations in Table 2. They show the changes in the averaged results obtained for the exposures of 1 and 24 h. The results for the Ni coating were also inserted for the comparison purpose; however, their interpretation is beyond the scope of this work and the following analysis will focus on the impedance behavior of the Ni–W samples. Resistance R 2 and parameters n 1 and Y 01 of the constant phase element CPE1 refer to the MF peak. R 2 did not change much with exposure time. The CPE1 parameter Y 01 increased with time, whereas the n 1 values became close to 0.9 (see Table 2) approximating the capacitor behavior. The values of the capacitance derived from the CPE1 parameters [37] were in the range 40–120 μF/cm2. This range is conceivable for the double-layer capacitance in aqueous solutions, especially when it is considered that a non-uniform dissolution of two-phase alloys can generate surface roughness. Therefore, the MF peak was related to the charge transfer process. Thus, the components of the equivalent circuit shown in Fig. 5 and corresponding to this phenomenon can be interpreted as the charge transfer resistance (R 2) and the constant phase element describing the double-layer capacitance (CPE1).
Another behavior was found by us for the resistance R 3 and the constant phase element CPE 2, which concern the LF response observed for the Ni–W coatings. Resistance R 3 increased with time, except for the Ni–11W sample, see Table 2. The estimated values of the CPE2 parameter Y 02 were very scattered and changed randomly with time, whereas the n 2 values were close to 0.6. The value 0.6 is higher than 0.5 that is characteristic for the diffusion impedance with semi-infinite diffusion layer (Warburg impedance). However, the shape of the LF response in the Nyquist format (either a flattened loop or a more-or-less linear tail turning to the real axis at low frequencies) suggests that the thickness of the diffusion layer is rather finite [38]. Under such conditions, the diffusion impedance is expressed by a complicated, hyperbolic tangent equation [38]. However, the finite-length diffusion through a thin, protective film can be approximated by a parallel combination of a resistor and a CPE element with parameter n close to 0.5 [39, 40]. It is conceivable that this situation can take place during the corrosion of Ni–W alloys due to the coverage of the surface with a thin film of corrosion products. Therefore, the parallel combination of the R 3 and CPE2 shown in Fig. 5 and describing the LF response could be related to the diffusion process through this surface film.
The analysis of the dependences of the impedance parameters on alloy composition (data in Table 2) yields a better insight into the corrosion process of the Ni–W coatings. Charge transfer resistance R 2 shows a decreasing trend with increasing W content in the alloy, see Fig. 6. This trend implies that the process is faster for the W-rich coatings. Such a behavior can be explained either by the activating effect of the grain refinement [1, 12] or by preferential dissolution of the W-rich phase [33]. On the other hand, the diffusion-related LF response was clearly pronounced for the W-rich alloys, see plots in Fig. 7, and the corresponding resistance, R 3, increased with time (data in Table 2) and W content, see Fig. 6. These findings inclined us towards a conclusion that the diffusion occurs through a thin-surface film of the corrosion products conceivably generated by the oxidation of W. The higher R 3 values for the W-rich coatings coincide with the faster dissolution (lower R 2) and, thus, with larger amounts of generated products of W oxidation. The surface film generated under these conditions could be thicker and/or more compact. In the case of tungstates, an inhibition effect can be also expected. This aspect is illustrated in Fig. 8. SEM morphology of the Ni–15W sample after the corrosion test indicated a severe attack along boundaries of grain colonies. This is associated with preferential dissolution of W segregations at these boundaries. The generated corrosion products formed a layer with a barrier or inhibition ability which could delay the corrosion process. For W-rich samples, the colony size decreased and the net of places of corrosion attack was more dense but the generated layer of corrosion products appeared to be more thick and/or dense. This matter will be addressed more deeply and documented by SEM and EDS data in a separate paper.

Effect of W content on resistances R 2, R 3 and R 2 + R 3

Comparison of impedance spectra taken after 24 h for Ni–W coatings with different W content (numbers at curves denote the W content)

SEM image of Ni–15W sample after corrosion test
The sum of R 2 and R 3 can be considered as an indicator of the corrosion resistance. This sum is smaller for the Ni–W coatings compared to that of the Ni sample, see Fig. 6, indicating that corrosion resistance of unalloyed Ni is higher. Interestingly, after a 24-h exposure, this sum is practically the same (within standard deviation) for all Ni–W compositions. This finding suggests that the corrosion resistance of all Ni alloys is very comparable irrespective of W content. As discussed above, this behavior can be a result of an interplay of the activating effect of grain refinement and of preferential dissolution of W-rich phase from one side and the diffusion barrier action and inhibition provided by the surface film of the W oxidation products from the other side.
The potentiodynamic polarization studies were performed after the completion of the second impedance measurement (after 27-h exposure). Typical polarization curves obtained for the studied alloy compositions are compared in Fig. 9.

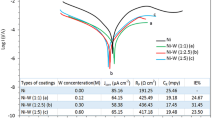
Anodic polarization curves for tested coatings in 0.5 M NaCl (numbers at curves denote W content)
An analysis of the polarization data in the vicinity of the corrosion potential allowed the determination of the corrosion parameters. Some selected results obtained from this analysis, namely: the corrosion potential (zero-current potential), E corr′; the corrosion current density, j corr; and the polarization resistance, R p, are presented in Table 3. E corr′ decreased with W content and coincided with those arising from Fig. 3. A comparison of the j corr values proved that the corrosion of the Ni–W alloy coatings proceeds much faster than of the Ni sample. The difference is rather large: of nearly one order of magnitude. The worse corrosion resistance of the Ni–W alloys can be ascribed to the activating effect of the grain refinement [1, 12] and to the two-phase structure of this alloy which favors the formation of galvanic couples and, thus, stimulates corrosion. Conceivably, these factors surpass the protective action of the W oxidation products.
It is interesting that all Ni–W coatings show comparable values of the corrosion current density. They remain in a very narrow band: from 3 to 4.3 μA/cm2, so there is no clear trend. A similar conclusion can be drawn from the results reported in work [1] on the Ni–W coatings deposited at 85 °C and containing 2–16% W. These findings contradict the results of work [9] done in a similar solution, where a significant decrease in j corr was observed with increasing W content from 5.5–16%. However, similar conclusions about quite comparable corrosion resistance of the tested Ni–W alloys can be made upon the respective polarization resistances, see Table 3. The values of R p are close to each other (the differences are within the standard deviation), except for the Ni–11W data. However, this sample data depart from the remaining Ni–W alloys also in other relations (a distinctive course of the anodic polarization curve seen in Fig. 9 and outlying data of impedance parameters with time placed in Table 2). This behavior can be related to the low-W content.
It is worth noting that the independence of the corrosion resistance of Ni–W alloy composition was also inferred from the impedance data. In Fig. 10, the resistance measures of the corrosion intensity derived from the polarization data (the polarization resistance R p) and from the impedance data (the sum of resistances R 2 + R 3) are compared. The compositional dependencies of those quantities confirm the inferior corrosion resistance of Ni–W vs. Ni. The slight effect of W content on the corrosion behavior of Ni–W alloys is also evident in each case. The resistance measures compared in Fig. 10 show very similar compositional dependences, but they have different values. The polarization tests were performed indirectly after the impedance measurement taken after 24 h exposure. However, they indicated that the R p values are two to three times less than the corresponding sums of the resistances obtained from these impedance measurements. This discrepancy can originate from the activating effect of the cathodic polarization and non-steady-state conditions prevailing under potentiodynamic measurements. It should be noted, that the readings of the corrosion potential, E corr′, determined from the polarization curves (given in Table 3) are slightly more negative, compared to the steady state values of E corr, see Fig. 3. Interestingly, the R p values approximate the sum R 2 + R 3 from the first impedance measurements taken in the initial period of the exposure, i.e., after 1 h, see Fig. 10.

Comparison of polarization resistance, R p, obtained from polarization measurements and the sum of resistances R 2 + R 3 determined from impedance measurements and taken after different exposure times: 1 and 24 h
Another point that deserves attention is the relation between corrosion current density and linear corrosion rate expressed in mm/year. This relation is given by the equation:
where: r is linear corrosion rate, F is the Faraday constant, M M and d M are atomic mass and density of metal M, n M is number of electrons exchanged in the reaction of metal dissolution, and k M is proportionality coefficient dependent on metal properties. Taking into consideration the relevant properties of Ni (atomic mass, 58.7 g/mol; number of exchanged electrons, 2; and density, 8.9 g/cm3) and W (atomic mass, 183.9 g/mol; number of exchanged electrons, 6—the oxidation to tungstate; and density, 19.3 g/cm3), it can be shown that the proportionality coefficient, k M, is ca. twice smaller for W than for Ni (0.016 and 0.034 mm3/C for W and Ni, respectively). This indicates that despite the identity of the j corr data, the linear corrosion rate should be smaller for W-rich alloys. The difference in the W content in the studied alloys is 10%. Assuming the stoichiometric dissolution of the studied Ni–W alloys, this reduction in r values is rather small; it equals ca. 5%—which is less than the standard deviations of j corr—see Table 3. However, this effect can be pronounced in the case of the preferential dissolution of W [33].
The course of the polarization curves seen in Fig. 9 indicates that there is a significant difference in the anodic behavior of the tested materials. The Ni coating shows an arrest in anodic current in the potential range from E corr till ca. 0.05 V. Then, an abrupt rise in the anodic current is observed which coincides with the onset of the pitting corrosion. The pitting became severe at higher anodic polarization. On the contrary, the Ni–W alloys exhibit more negative corrosion potentials and higher cathodic and anodic currents in the vicinity of their E corr. The anodic polarization of these alloys leads to a gradual increase in the dissolution current till a maximum is reached at ca. −0.1 to +0.2 V and then, after a slight decrease, a wide plateau is observed. A similar shape of the anodic curves was reported for Ni–W [30] and Co–W electrodeposits [42] in NaCl solutions. It is worth noting that this suppression of the dissolution of Ni–W coatings occurs in the region of a sharp increase of anodic current and intense dissolution of the pure Ni sample. This behavior can be associated with the inhibiting action of the formed W oxidation products [41]. Tungstates form a cation-selective film which resists the incorporation of the chloride anions at higher potentials and, thus, withstands the pitting [41]. Of interest is that the course of the polarization curves for all Ni–W coatings is quite similar at high positive potentials, see Fig. 9.
The dissimilarity in the dissolution behavior of Ni–W and unalloyed Ni in NaCl solution with increasing anodic potential is clearly seen in Fig. 11. In this diagram, anodic current densities for the Ni and Ni–W samples are compared at two selected anodic potentials. The first selected potential, E = 0 V, lies in the region of dissolution suppression for Ni, whereas the second potential, E = 0.25 V, exceeds the breakdown potential for Ni and causes severe pitting. At the lower potential, the values of j a for the Ni–W alloys are about two orders of magnitude higher than for unalloyed Ni. The situation changes dramatically at 0.25 V. Under these conditions, the values of j a for the Ni–W coatings are about one order of magnitude lower than for pure Ni. Moreover, the anodic current densities for Ni–W alloys (except for the Ni–11W sample) are practically the same at both potentials.

Effect of W content on anodic current density of Ni–W alloys at potentials E = 0 and 0.25 V
After the completion of the corrosion studies the morphology of the tested coatings was inspected with a light microscope. A significant difference in the surface state was revealed. A micrograph obtained for the Ni coating and presented in Fig. 12a shows a severe pitting. The lateral size of the pits approaches 100 μm. Completely different surface damages were observed for the Ni–W coatings. Their surface exhibited iridescent colors, characteristic for W oxidation products, either on the entire sample surface (see Figs. 12b,d, and e) or on parts of it (see Fig. 12c and f). Conceivably, these oxidation products can generate the diffusion restriction for the corrosion process what was observed in the impedance test. Samples Ni–11W (Fig. 12b) and Ni–20W (Fig. 12e) are quite uniformly corroded, whereas sample Ni–18W (Fig. 12d) shows extensive localized attack along cracks. Slightly marked, tiny lines of cracks were already observed for some samples at the time of receiving. These cracks were made deeper by the corrosion process. The corrosion damages for the Ni–15W (Fig. 12c) and Ni–21W samples (Fig. 12f) occur only locally, but they appear close to the cracks and especially the points of their convergence. It is obvious, that these damages were intensified upon anodic polarization. However, it is conceivable that this localized corrosion attack was initiated during the long exposure under the open circuit conditions and it corresponds to the corrosion characteristics presented in Tables 2 and 3.

Micrographs of samples surface after the corrosion test: a Ni, b Ni–11W, c Ni–15W, d Ni–18W, e Ni–20W and f Ni–21W
In this work, a rather big set of Ni–W samples was studied. The micrographs presented in Fig. 12 are appropriately selected to show the most representative images of the corrosion damages for the studied Ni–W compositions. However, these results do not allow us to make a definite conclusion on the effect of the alloy composition on the corrosion induced morphology; neither the influence of the coatings thickness is clear.
Despite obvious structural differences between the low-W and high-W alloys, the mode of corrosion damage did not coincide much with the alloy microstructure and the transition from the microcrystalline to the amorphous state. Quite different pictures (more uniform corrosion or intense attack at cracks) were observed within the same composition of the samples. These differences are rather more related to the preparation conditions than to the alloy composition. Various corrosion damages were observed for the samples of very similar composition and prepared from the same solution in consecutive experiments (with various charge passed). However, the difference in the corrosion resistance of the Ni–W coatings with vast cracks and without visible cracks was much smaller (within reported standard deviations) than that described in paper [34]. This point requires further studies.
Conclusions
The samples of the Ni–W coatings that were taken for the investigation contained from 11 to 21 at.% W, were of 7–52 μm thickness, and were electrodeposited from the citrate solution with different cathodic current density and deposition time. These coatings were divided into five groups of similar composition and used for structural and corrosion tests.
The XRD studies of the low-W-content coatings revealed the presence of two phases: a solid solution of W in fcc Ni and a new one, not described until now, constituted by a solid solution of Ni in bcc W. The increase in W content in the Ni–W coatings resulted in the grain refinement and the transition from the microcrystalline to amorphous/nanocrystalline structure at ca. 20% W.
The Ni–W alloys, in NaCl solutions, exhibited a clearly decreased corrosion resistance compared to the pure Ni coating, while all tested Ni–W coatings showed a quite comparable corrosion resistance in this solution. No clear relation between corrosion rate and W content was detected. This behavior can be a result of the interplay of the activating effect of the grain refinement and the preferential dissolution of W from one side and the diffusion barrier action or the inhibition provided by the surface film of W oxidation products from the other side.
The studied Ni–W coatings exhibited different corrosion damages: from quite uniform corrosion to localized attack at the cracks. These differences are rather more related to the preparation conditions than to the alloy composition.
An inversion in the dissolution behavior of Ni–W and unalloyed Ni was observed with increasing anodic potential. Contrary to pure Ni, the Ni–W coatings were resistant to pitting corrosion in NaCl solution.
References
Sriraman KR, Ganesh Sundara Raman S, Seshadri SK (2007) Mater Sci Eng A 460–461:39 doi:10.1016/j.msea.2007.02.055
Eliaz N, Sridhar TM, Gileadi E (2005) Electrochim Acta 50:2893 doi:10.1016/j.electacta.2004.11.038
Pisarek M, Janik-Czachor M, Donten M (2008) Surf Coat Tech 202:1980 doi:10.1016/j.surfcoat.2007.08.047
Obradovic M, Stevanovic J, Despic A, Stevanovic R, Stoch J (2001) J Serb Chem Soc 66(11–12):899
Ren R, Wu YC, Shu X, Shi CW, Li Y, Zheng YC (2005) Trans Nonferous Met Soc China 15:198, in Chinese
Donten M, Stojek Z, Królikowski A, Płonska E (2007) 211th Meeting of the Electrochemical Society, Chicago, Abs 549
Lee SL, Lee YF, Chang MH, Lin JC (1999) Corros Prev Contr 46(3):71
Stepanova LI, Purovskaya OG (1998) Met Finish 96:50 doi:10.1016/S0026-0576(98)80871-4
Ke ST, Lee JL, Hou KH, Ger MD (2006) J Technol 21:75, in Chinese
Atanassov N, Gencheva K, Bratova M (1997) Plat Surf Finish 84:67
Zhu L, Zhong Q, Liu J (2000) Plat Surf Finish 87:74
Królikowski A (2007) Ochr p Kor (in Polish) 50(4):140
Yao S, Zhao S, Guo H, Kowaka M (1996) Corrosion 52(3):183
Yamasaki T, Schlossmacher P, Erlich K, Ogino Y (1998) Nanostruct Mater 10(3):375 doi:10.1016/S0965-9773(98)00078-6
Galikova Z, Chovancova M, Danielik V (2006) Chem Pap 60(5):353 doi:10.2478/s11696-006-0064-2
Wu Y, Chang D, Kim D, Kwon S (2003) Surf Coat Tech 162:269 doi:10.1016/S0257-8972(02)00699-0
Wu Y, Chang D, Kim D, Kwon S (2003) Surf Coat Tech 173:259 doi:10.1016/S0257-8972(03)00449-3
Yang FZ, Guo YF, Huang L, Xu SK, Zhou SM (2004) Chin J Chem 2:228
Nawarro-Flores E, Chong Z, Omanovic S (2005) J Mol Catal Chem 226:179 doi:10.1016/j.molcata.2004.10.029
Somekawa T, Nieh TG, Higashi K (2004) Scr Mater 50:1561
Nasu T, Sakurai M, Kamiyama T, Usuki T, Uemura O, Yamasaki T (2002) J Non-Cryst Solids 312–314:319 doi:10.1016/S0022-3093(02)01702-7
Schuh CA, Nieh TG, Iwasaki H (2003) Acta Mater 51:431 doi:10.1016/S1359-6454(02)00427-5
Giga A, Kimoto Y, Takigawa Y, Higashi K (2006) Scr Mater 55:143 doi:10.1016/j.scriptamat.2006.03.047
Sulitanu N, Brinza F (2003) J Optoelectronics. Adv Mater 5(2):421
Yamasaki T (2000) Mater Phys Mech 1:127
Itoh K, Wang F, Watanabe T (2001) Nippon Kinzoku Gakkaishi 65(11):1023 in Japanese
Donten M, Cesiulis H, Stojek Z (2000) Electrochim Acta 45:3389 doi:10.1016/S0013-4686(00)00437-0
Donten M, Stojek Z (1996) J Appl Electrochem 26:665 doi:10.1007/BF00253466
Cesiulis H, Baltutiene A, Donten M, Donten ML, Stojek Z (2002) J Solid State Electrochem 6:237 doi:10.1007/s100080100225
Wang H, Yao S, Matsumura S (2002) Surf Coat Tech 157:166 doi:10.1016/S0257-8972(02)00151-2
Moussa SO, Ibrahim MAM, Abd El Rehim SS (2006) J Appl Electrochem 36:333 doi:10.1007/s10800-005-9069-8
Zhu L, Younes O, Ashkenasy N, Schacham-Diamand Y, Gileadi E (2002) Appl Surf Sci 200(1–4):1 doi:10.1016/S0169-4332(02)00894-2
Zhang L, Macdonald DD (1998) Electrochim Acta 43(18):2661 doi:10.1016/S0013-4686(97)00268-5
Sakai Y, Shitanda I, Itagaki M, Watanabe K, Yasuda K, Saitou M (2007) 7th Intern Symp on Electrochemical Impedance Spectroscopy, Argeles-sur-Mer, Abstract 96
Magalhaes AAO, Margarit ICP, Mattos OR (1999) Electrochim Acta 44:4281 doi:10.1016/S0013-4686(99)00143-7
Magalhaes AAO, Margarit ICP, Mattos OR (2004) J Electroanal Chem 572(2):433 doi:10.1016/j.jelechem.2004.07.016
Hsu CS, Mansfeld F (2001) Corrosion 57(9):747
Jacobsen T, West K (1995) Electrochim Acta 40:255 doi:10.1016/0013-4686(94)E0192-3
Campestrini P, van Westing EPM, Hovestad A, de Wit JHW (2002) Electrochim Acta 47:1097 doi:10.1016/S0013-4686(01)00818-0
Campestrini P, Terryn H, Vereecken J, de Wit JHW (2004) J Electrochem Soc 151(6):B370 doi:10.1149/1.1736683
Lloyd AC, Noel JJ, McIntyre S, Shoesmith DW (2004) Electrochim Acta 49:3015 doi:10.1016/j.electacta.2004.01.061
Zhang YZ, Yao M (1999) Trans IMF 77(2):78
Author information
Authors and Affiliations
Corresponding author
Additional information
Contribution to the Fall Meeting of the European Materials Research Society, Symposium D: 9th International Symposium on Electrochemical/Chemical Reactivity of Metastable Materials, Warsaw, 17th–21st September, 2007.
Rights and permissions
About this article
Cite this article
Królikowski, A., Płońska, E., Ostrowski, A. et al. Effects of compositional and structural features on corrosion behavior of nickel–tungsten alloys. J Solid State Electrochem 13, 263–275 (2009). https://doi.org/10.1007/s10008-008-0712-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-008-0712-2