
Abstract
Cobalt-molybdenum coatings were prepared by electrodeposition in a sulfate-citrate bath and their morphology, structure and magnetic properties were analysed. Concentrations of 0.1 mol dm−3 CoSO4 and 0.005 mol dm−3 Na2MoO4 at pH 6.6 led to Co-Mo deposits of 20–23% Mo that can be grown to several microns over graphite or copper substrates. At low deposition potentials or current densities, the deposits presented a close-packed hexagonal structure (hcp) that evolved to a (100)+(110) preferred orientation and acicular morphology as the deposit thickness increased. When the deposition potential or the current density was made more negative, a mixed crystalline+amorphous structure was obtained. The degree of crystallinity depended on the thickness: thin films were more amorphous than the thicker ones. Co-Mo deposits showed lower saturation magnetization (M s) and coercivity (H c) than the pure cobalt deposits. The crystalline+amorphous films showed the lowest H c values (around 40 Oe).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Magnetic materials of iron group metals have wide applications in industry, mainly in micro-electromechanical systems (MEMS) and magnetic storage devices. The presence of molybdenum modifies their magnetic properties [1, 2] and improves their hardness and corrosion resistance [3, 4, 5]. These new properties make iron group metal-molybdenum alloys promising materials for microelectronic applications.
Interest in the electrodeposition of molybdenum with iron group metals has thus increased. Codeposition of molybdenum with iron group metals is induced, since complete reduction of molybdenum in aqueous solutions does not occur in the absence of an iron group metal [6]. Several studies have examined the anomalous electrodeposition of Ni-Mo [7, 8, 9, 10, 11, 12, 13, 14, 15], especially in a citrate medium, although there are fewer reports on Co-Mo or Fe-Co deposition [16, 17, 18].
In a previous study [2] the Co-Mo electrodeposition from a sulfate-citrate bath was explored. The percentage of Mo in the deposits was up to 60%, although the films were highly stressed. It was claimed that the presence of Mo led to soft magnetic behaviour.
A more recent study of the influence of bath composition and pH on the electrochemical behaviour of Co-Mo deposition suggested that the equilibria between the species in solution played an important role. It was found that alloy deposition occurred through different stages depending on the pH of the bath, which affected the composition of the final deposit [19]. We fixed the working pH at 6.6 to decrease simultaneous hydrogen evolution. In the present study, bath composition was fixed at 0.1 mol dm−3 cobalt sulfate and 0.005 mol dm−3 sodium molybdate. Citrate concentration was adjusted at 0.2 mol dm−3 in order to complex cobalt(II); higher citrate concentrations hinder the codeposition and decrease the efficiency of the process [19]. Molybdate concentration was kept low to prevent large Mo incorporation in the deposits, which would decrease the saturation magnetization of the coating.
The aim of the present study was to examine the influence of various electrodeposition parameters on the properties of the deposit at this fixed bath composition. We tested the deposition over a range of substrates in order to improve the film adherence and we analysed the influence of the thickness and the electrodeposition conditions on structure and magnetic properties, since our main interest was the preparation of homogeneous films with soft magnetic behaviour.
Experimental
The electrochemical measurements were performed in a conventional three-electrode cell using a microcomputer-controlled Autolab potentiostat/galvanostat with PGSTAT30 equipment and GPES software. All chemicals used were of analytical grade. Solutions were freshly prepared with water that was first doubly distilled and then treated with a Millipore Milli-Q system. Before and during the experiments, solutions were de-aerated with argon. The temperature was maintained at 25 °C.
The morphology of the deposits was examined with a Hitachi S 2300 scanning electron microscope. Elemental composition was determined with an X-ray analyser incorporated in a Leica Cambridge Stereoscan S-360 scanning electron microscope. The efficiency of deposition was calculated by comparing the reduction charge and the results of chemical analysis of the films. For chemical analysis, the deposits were dissolved in 5 mL of 1% nitric acid solution and samples were analysed by inductively coupled plasma mass spectrometry (ICP-MS). Measurements were made in a Perkin-Elmer spectrometer Elan 6000. Certified standard solutions of cobalt and molybdate ions containing rhodium as an internal standard were used to calibrate the instrument.
The thickness of the deposits was measured on a Zeiss Axiovert 405M microscope.
X-ray diffraction (XRD) phase analysis was performed in a Philips MRD diffractometer in low-resolution parallel beam optics. The Cu-Kα radiation (λ=1.5418 Å) was selected by means of a diffracted-beam flat graphite monochromator. Diffractograms were obtained in the 20–100° 2θ range with a step of 0.05° and a measuring time of 5″ per step. The magnetic measurements were performed with a Manics DSM8 pendulum-type magnetometer at room temperature or with a SQUID magnetometer.
Vitreous carbon (r=1.0 mm), graphite (r=4.1 mm) and copper (r=1.0 mm) rods were used as working electrodes. The vitreous carbon electrode was polished to a mirror finish before each experiment using alumina of different grades (3.75 and 1.85 μm) and cleaned ultrasonically for 2 min in water. Copper and graphite electrodes were also polished before each experiment using sandpaper (2400 and 4000) and, later, damp alumina (0.3 μm). The reference electrode was an Ag/AgCl/1 mol dm–3 NaCl electrode mounted in a Luggin capillary containing 0.5 mol dm–3 Na2SO4 solution. All potentials are referred to this electrode. The counter electrode was a platinum spiral.
During electrodeposition the solution was gently stirred (ω=60 rpm) to avoid the depletion of the minority species (molybdate) to the electrode and to ensure homogeneous composition of the deposits.
Results and discussion
Electrochemical study
The general behaviour of Co-Mo deposition was analysed on vitreous carbon, graphite and copper substrates to identify the best electrode for thick-adherent coatings. Voltammetric, chronoamperometric and chronopotentiometric studies were carried out for this purpose.
The redox process was similar in all substrates, but the electrodes behaved differently in the reduction (Fig. 1). Copper was the most active substrate and vitreous carbon was the slowest. Alloy deposition started at around −900 mV for copper, at −1050 mV for graphite and at −1150 mV for the vitreous carbon electrode. A complex oxidation peak was observed in all cases, at around −200 mV. According to this, it seemed that alloy deposits could be obtained in a similar way over the different substrates, although the potential necessary to start the deposition process was different for each electrode.

Cyclic voltammograms of a 0.1 mol dm–3 CoSO4+0.2 mol dm–3 Na3C6H5O7+0.005 mol dm–3 Na2MoO4, pH 6.6, solution (Co-Mo solution): (a) copper, (b) graphite and (c) vitreous carbon electrodes; scan rate 50 mV s−1
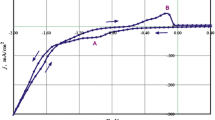
To test the adherence of Co-Mo deposits on those substrates, various samples were prepared at long deposition times. When deposits were obtained potentiostatically on the vitreous carbon electrode, there were deformations in the j–t curves in spite of stirring (Fig. 2A) and the films were cracked (Fig. 2B). At longer deposition times the deposits fell off the substrate (Fig. 2C). Vitreous carbon was therefore ruled out. Graphite or copper substrates were suitable because the deposits did not detach even for thick coatings.

j–t transient from −500 mV to −980 mV; 60 rpm, vitreous carbon electrode, Co-Mo solution. B, C SEM images of the deposits obtained at points a and b of A, respectively
Characterization of the cobalt-molybdenum deposits
According to the results earlier obtained, Co-Mo deposits of several microns were prepared on graphite or copper electrodes. Morphological, compositional and structural characterization was carried out for each sample. Our main objective was focused on controlling the structure of the deposits, because it is known that structure and magnetic properties are usually related. Graphite electrodes were more useful because their greater area improved the resolution of the response.
Pure cobalt deposits were prepared from a molybdate-free bath in order to compare them with the Co-Mo deposits. Potentials or current densities were chosen to give similar deposition rates.
Figure 3 shows the diffractograms of cobalt and cobalt-molybdenum (~23% Mo) deposits obtained at a similar deposition rate (j=−3.2 mA cm−2) and the corresponding chronopotentiometric curves. The potential related to pure cobalt deposition evolved to a more negative value than that corresponding to Co-Mo deposition (Fig. 3A), because alloy deposition was favoured [2]. The diffractograms showed narrow peaks, revealing the crystalline nature of the deposits (Fig. 3B). For pure cobalt, the main peaks corresponded to a close-packed hexagonal structure (hcp), whereas a small peak around 2θ=51.5° is attributed to face-centred cubic (fcc) cobalt deposited next to the main hcp structure.

E–t transients recorded applying −3.2 mA cm−2, graphite electrode, 60 rpm: (a) cobalt deposit obtained from a 0.1 mol dm–3 CoSO4+0.2 mol dm–3 Na3C6H5O7, pH 6.6, solution; (b) Co-Mo deposit of 23% Mo obtained from the Co-Mo solution. B X-ray diffractograms of the deposits from A; 140 C cm−2, 32 μm
The diffractogram of a Co-Mo deposit shows a pure hcp structure, although the position and relative height of the peaks are different from the pure cobalt deposit. The peaks of the hexagonal structure were shifted to lower values corresponding to an increase of the cell parameters, as a consequence of the incorporation of molybdenum. The cell parameters of this hexagonal structure were calculated after fit profile and least-squares refinement, leading to values of a=2.550 Å and c=4.125 Å, greater than those for pure cobalt (a=2.507 Å, c=4.092 Å). Moreover, the Co-Mo deposit showed greater orientation, the hk0 being the main diffraction peaks. The practical absence of peaks with l≠0 indicated that the [001] crystallographic direction was nearly parallel to the substrate surface.
In a previous study, thinner (0.2 μm) Co-Mo deposits with an amorphous structure had been obtained [2]. In contrast, Co-Mo deposits of several microns prepared in the present work had a crystalline structure. Since the former were prepared potentiostatically and using a vitreous carbon electrode, we explored the influence of deposition parameters on the structure of the Co-Mo coatings.
Influence of deposition technique and the substrate nature
For all the conditions tested, potentiostatic and galvanostatic techniques led to equivalent electrochemical parameters: when a current density value j 1 was applied, the potential reached a stationary value E 1; analogously, when E 1 was the potential value applied, the stationary current recorded from the j–t transient was j 1. Moreover, by comparing the deposits obtained with both techniques, the same morphology and composition were found.
For example, irrespective of the technique, thick Co-Mo deposits corresponded to an acicular morphology when a graphite substrate was used (Fig. 4). The process was highly efficient for these deposits (around 85%).

SEM image of a deposit obtained at −940 mV during 55,000 s; 23% Mo, 32 μm,Co-Mo solution, 60 rpm, graphite electrode
On the other hand, the galvanostatic transients of Co-Mo deposition over graphite and copper electrodes at the same current density (−3.2 mA cm−2) showed that practically the same stabilization potential was attained in both cases. The deposits obtained on copper substrate at long deposition times showed the same morphology and composition as those obtained over the graphite electrode.
When the diffractograms of the thick Co-Mo deposits obtained through the potentiostatic/galvanostatic technique and over the copper/graphite substrate were compared, similar profiles were also observed (Fig. 5). An hcp structure with hk0 main peaks was always obtained. The position of these peaks did not change, as corresponds to a given value of the molybdenum percentage. Similar results were obtained when potentials lower than −1000 mV were used.

X-ray diffractograms of the deposits obtained at −3.2 mA cm−2 from the Co-Mo solution; 60 rpm: (a) copper electrode, (b) graphite electrode
Influence of the deposit thickness
When deposits obtained at different deposition times for a given potential value were analysed, it was corroborated that the composition of the coatings did not change with the deposition time, due to the stirring conditions. However, the morphology and structure presented some differences as a function of the reduction charge. Thick Co-Mo coatings had the morphology shown in Fig. 4, while thinner coatings presented the morphology shown in Fig. 6A. Figure 6B shows the corresponding X-ray diffractograms. A relationship between the change in the morphology and the structure was observed. As the thickness of the deposit increased, the hcp structure increased its preferred orientation, leading to a structure with a clear (100)+(110) preferred orientation. At the same time, the morphology changed from a polyhedral grain to acicular.

SEM micrograph of the deposits obtained at −940 mV from the Co-Mo solution; 60 rpm, graphite electrode, 8 μm. B X-ray diffractograms of Co-Mo deposits with (a) 8 μm and (b) 32 μm
Influence of the deposition potential
The influence of the deposition rate on the morphology and structure of the coatings was analysed by means of the variation of the deposition potential, because the deposition rate increases as the applied potential is more negative. In the potential range studied, the applied potential had little effect on the composition (Table 1). However, a clear decrease of the deposit thickness was obtained by making more negative the deposition potential as a consequence of the lowering of the efficiency (η).
The morphology, composition and crystalline structure of the deposits obtained at a fixed charge (20 C cm−2) were studied. Deformations in the j–t transients were observed for potentials equal to or more negative than −1000 mV (Fig. 7A), probably attributable to simultaneous hydrogen evolution. At deposition potentials less negative than −1000 mV, non-cracked coatings were obtained (Fig. 7B, image a), showing the previously described polyhedral morphology for thin Co-Mo films. In contrast, stressed and cracked deposits were obtained from −1000 mV (Fig. 7B, images b and c). Diffractograms of deposits obtained at different potentials showed some differences, as can be observed in the magnified X-ray diffractograms in the range 35–60° 2θ (Fig. 7C). For the deposits obtained at −940 mV, the narrow 100, 002 and 101 peaks of the hexagonal structure can be observed next to small peaks of the substrate. For those obtained at −1000 mV, there were small 100 and 101 peaks corresponding to a hexagonal crystalline structure, peaks corresponding to a graphite substrate and a very wide peak (3.5° 2θ) centred on 44.5° 2θ and attributed to an amorphous Co-Mo phase [2]. Thus, at this potential, the deposit seems to have a mixed crystalline and amorphous structure. The diffractogram of the deposit obtained at −1100 mV mainly showed peaks of the substrate and a wide peak attributed to the amorphous phase. Thus, a decrease of the deposition potential, i.e. an increase of the deposition rate, favoured the formation of less crystalline deposits. For the crystalline+amorphous samples, the presence of cracks or a non-compact structure allowed the observation of the substrate peaks in the diffractogram.

j–t transients of the Co-Mo solution; 60 rpm, graphite electrode: (a) −940, (b) −1000, (c) −1100 mV. B SEM images of the deposits obtained from A; 20–23% Mo, 20 C cm−2: (a) 4.6 μm, (b) 2.7 μm, (c) 0.8 μm. C X-ray diffractograms of these deposits
At higher deposition charges of around 150 C cm−2, for which deposits of several microns were obtained, XRD revealed the formation of crystalline deposits at −940 and −1000 mV. On the other hand, a mixture of crystalline+amorphous phases was detected at −1100 mV, since the 100 peak of the hcp structure and the wide peak attributed to the amorphous structure were detected (Fig. 8). This result evidenced that, at the intermediate deposition potential (−1000 mV), the crystallinity increased with the deposit thickness. In contrast, for the deposits prepared at E<−1000 mV, the structure was not affected by the deposit thickness.

X-ray diffractograms of deposits obtained under the same conditions as in Fig. 7 but with a greater charge: (a) −940, (b) −1000, (c) −1100 mV; 150 C cm−2; (a) 35 μm, (b) 20 μm, (c) 6.5 μm
Magnetic behaviour of Co-Mo films
Since different Co-Mo structures were observed as a function of the deposition potential (or current density), deposits of several microns were prepared to analyse their magnetic properties. A graphite electrode and high thicknesses were selected to facilitate separation of the coatings from the substrate.
The magnetic properties were determined by recording hysteresis loops (Fig. 9). The easy axis is the in-plane direction of the parallel film. For a parallel applied field (H ∥) a large saturation field (H s∥) of about 9 kOe was required to align the magnetization in the film plane and a low remanence value was observed. The results obtained under a perpendicular field (H ⊥) were similar, except that the saturation field (H s⊥) was about 20 kOe. These features indicated strong uniaxial anisotropy, in addition to the usual shape anisotropy [20].

Parallel and perpendicular hysteresis loops for a deposit obtained from Co-Mo solution; 60 rpm, graphite electrode, −3.2 mA cm−2, 22% Mo, 40 μm
The value of the uniaxial anisotropy constant (K) and the angle θ of the resulting anisotropy with respect to the film normal may be found using the expressions [21]:
and:
where M s is the saturation magnetization. Values of K≈6.4×106erg cm−3 and θ=55° were found.
A strong uniaxial anisotropy was observed for the Co-Mo samples, as expected for an hcp structure and high textured films. However, the K value obtained was greater than that corresponding to pure hcp cobalt (K=4.5×106 emu cm−3) [20], probably due to the residual stress and distortions caused by the presence of molybdenum in the crystalline lattice of cobalt.
The magnetization curves of Co-Mo deposits were compared with those corresponding to pure cobalt deposits obtained from a molybdate-free solution and prepared at similar deposition rates (Fig. 10). For the pure cobalt coating with an hcp structure, a saturation magnetization of 135 emu g−1 was obtained, which corresponded to a bulk cobalt response. The coercivity value (H c) was around 160 Oe.

Magnetization versus magnetic field applied for the deposits obtained at 60 rpm over a graphite electrode: (a) 0.1 mol dm–3 CoSO4+0.2 mol dm–3 Na3C6H5O7, pH 6.6 solution, −1120 mV, 37 μm; (b) Co-Mo solution, −940 mV, 35 μm; (c) Co-Mo solution, −1100 mV, 6.5 μm
For cobalt-molybdenum deposits, a clear decrease of the saturation magnetization was observed, around 65 emu g−1. This value was approximately constant for deposits prepared at different deposition rates, because they contained similar molybdenum percentages.
Moreover, the introduction of molybdenum in the deposits caused a clear decrease of the coercivity of the coatings. The Co-Mo films were soft, with coercivities in the range 70–40 Oe. The deposits obtained at potentials no more negative than −1000 mV (or at current densities no more negative than −5 mA cm−2) showed similar values of coercivity (around 70 Oe). However, deposits prepared at more negative values of the current density or the potential (until −1200 mV) showed a lower coercivity (around 40 Oe), revealing that amorphous+crystalline coatings presented the lowest value of any of these films.
Conclusions
Electrodeposition is a suitable way to obtain Co-Mo magnetic deposits over different substrates from a sulfate-citrate bath at pH 6.6. As deduced from voltammetric experiments, the start of the alloy deposition over vitreous carbon is delayed with respect to graphite and copper electrodes. Copper and graphite are useful to prepare Co-Mo coatings of several microns, whereas vitreous carbon favours detachment of the films.
The bath used, although it contains a low concentration of molybdate, leads to cobalt-molybdenum deposits of 20–23% Mo. This percentage is slightly dependent on the deposition potential or the current density applied. It is possible to maintain a constant composition through the thickness of the coating using moderate stirring of the solution during the deposition. No influence of the substrate (copper/graphite) or the preparation technique has been detected on the properties of the Co-Mo films. Co-Mo coatings of an hcp structure have been always obtained, and the cell parameters are clearly greater than those for the hexagonal cobalt. The films evolve with thickness to a structure with a clear (100)+(110) preferred orientation, showing an acicular morphology.
However, the properties (morphology, structure and magnetic properties) of the cobalt-molybdenum coatings are dependent on the deposition rate. The incorporation of molybdenum in the cobalt coatings leads to a decrease of both saturation magnetization and coercivity, with respect to the values for pure cobalt. The value of the saturation magnetization was constant for a fixed molybdenum percentage, but the value of the coercivity was also dependent on the crystalline structure of the films. Low deposition rates (low deposition potentials or current densities) favoured the formation of homogeneous crystalline deposits with a coercivity approximately constant. However, when high deposition rates were used to prepare the films, cracked coatings were obtained, with a mixed crystalline+amorphous structure and a lower coercivity than those for pure crystalline ones.
For the solution studied, the selection of both coating thickness and deposition potential (or current density) allows us to control the crystalline nature of the Co-Mo deposits. A decrease of the crystallinity improves the soft magnetic behaviour of the Co-Mo coatings.
References
Taylor WP, Schneider M, Baltes H, Allen MG (1997) In: Proceedings of the international conference on solid-state sensors and actuators, Transducers’97, Chicago. Transducer, Chicago, pp 1445
Gómez E, Pellicer E, Vallés E (2001) J Electroanal Chem 517:109
Kinh VQ, Chassaing E, Saurat M (1975) Electrodep Surf Treat 3:205
Friend WZ (1980) Corrosion of nickel and nickel alloys. Wiley-Interscience, New York, pp 95–135, 248
Yao SW, Zeng Y, Guo HT (1994) Surf Tech (Japan) 45:643
A. Brenner (1963) Electrodeposition of alloys, vols 1–2. Academic Press, New York
Nee CC, Kim W, Weil R (1988) J Electrochem Soc 135:1100
Chassaing E, Vu Quang K, Wiart R (1989) J Appl Electrochem 19:839
Crousier J, Eyraud M, Crousier JP, Roman JM (1992) J Appl Electrochem 22:749
Chassaing E, Roumegas MP, Trichet MF (1995) J Appl Electrochem 25:667
Podhala EJ, Landolt D (1996) J Electrochem Soc 143:885
Podhala EJ, Landolt D (1996) J Electrochem Soc 143:893
Hu CC, Weng CY (2000) J Appl Electrochem 30:499
Murase K, Ando H, Matsubara E, Hirato T, Awakura Y (2000) J Electrochem Soc 147:2210
Zeng Y, Li Z, Ha M, Zhen S (2000) Electrochem Commun 2:36
Beltowska-Lehman E (1990) J Appl Electrochem 20:132
Podhala EJ, Landolt D (1997) J Electrochem Soc 144:1672
Niu ZJ, Yao SB, Zhou SM (1998) J Electroanal Chem 455:205
Gómez E, Pellicer E, Vallés E (2003) J Electroanal Chem 556:137
Klavunde KJ (2001) Nanoscale materials in chemistry. Wiley-Interscience, New York
Xiao JQ, Chien CL, Gavrin A (1996) J Appl Phys 79:5309
Acknowledgements
The authors thank the Serveis Cientificotècnics (Universitat de Barcelona) for the use of their equipment, the Servei de Magnetoquímica (Universitat de Barcelona) and Dr B. Martínez of Institut de Ciència de Materials de Barcelona (ICMAB) for magnetic measurements. This paper was supported financially by contract MAT 2000-0986 from the Comisión Interministerial de Ciencia y Tecnología (CICYT) and by the Comissionat of the Generalitat de Catalunya under Research Project 2001 SGR 00046. E.P. also thanks the DURSI of the Generalitat de Catalunya for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gómez, E., Pellicer, E., Alcobé, X. et al. Properties of Co-Mo coatings obtained by electrodeposition at pH 6.6. J Solid State Electrochem 8, 497–504 (2004). https://doi.org/10.1007/s10008-004-0495-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-004-0495-z