
Abstract
Three fully amorphous comb-branch polymers based on poly(styrene-co-maleic anhydride) as a backbone and poly(ethylene glycol) methyl ether of different molecular weights as side chains were synthesized. SiO2 nanoparticles of various contents and the salt LiCF3SO3 were added to these comb-branch polymers to obtain nanocomposite polymer electrolytes. The thermal and transport properties of the samples have been characterized. The maximum conductivity of 2.8×10−4 S cm−1 is obtained at 28 °C. In the system the longer side chain of the comb-branch polymer electrolyte increases in ionic conductivity after the addition of nanoparticles. To account for the role of the ceramic fillers in the nanocomposite polymer electrolyte, a model based on a fully amorphous comb-branch polymer matrix in enhancing transport properties of Li+ ions is proposed.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
After Wright and co-workers [1] discovered the ionic conductivity in alkali metal salt complexes of poly(ethylene oxide) (PEO) in 1973, solid polymer electrolytes have received considerable attention because of their potential in various electrochemical applications, such as lithium batteries, sensors, and electrochromic and photoelectrochemical devices [2, 3]. PEO contains ether coordination sites, which assist the dissociation of salts incorporated in the polymer, as well as a flexible macromolecular chain structure for promoting facile ionic transport. However, polymer electrolytes based on a PEO matrix show comparatively lower ionic conductivity at ambient temperature, the reason being the existence of crystalline domains while ion carriers can only transfer within its amorphous phase [4].
In order to enhance the conductivity, several approaches have been suggested in the literature [5, 6]. At first an organic plasticizer, such as ethylene carbonate, propylene carbonate or tetraethylene glycol, etc., was added to the polymer electrolyte. The conductivities in these systems are higher than 10−4 S cm−1 at ambient temperature. Unfortunately, the plasticizer easily results in electrolyte leakage and deteriorates the polymer electrolyte’s mechanical properties so as to lose the connection between the electrode and electrolyte. These drawbacks lead to serious problems in terms of battery cyclability and safety hazards. High ionic conductivity and good electrochemical and chemical stabilities are of crucial importance to polymer electrolytes; indeed, only a dry polymer electrolyte can ensure an efficient cyclability of the lithium metal electrode, as clearly confirmed by a recent paper [7]. The addition of inorganic fillers for enhancing the mechanical stability and conductivity is an important strategy. A polymer electrolyte consisting of semicrystalline PEO and a non-conducting ceramics filler has been studied, and an increase in conductivity with the addition of small amounts of insulating filler (less than 10%) was found in some cases [8, 9, 10, 11]. Some mechanisms have been proposed to account for the increase in conductivity: a highly conductive layer forms at the polymer/ceramic interface, or there is a Lewis acid–base interaction between Lewis acid sites or the OH– group on the nanoparticle surface and both the ClO4 − anions and the PEO segments, or that the ceramic filler merely prevents the PEO from crystallizing effectively [9, 10, 11]. With the addition of the inorganic filler, the key feature of previous works used a semicrystalline PEO matrix and showed that the conductivity of the polymer electrolyte was lower than 10−5 S cm−1, but this is still not suitable for application in a lithium battery. To improve the ambient conductivity, the polymer has been used in modified forms, such as in polymer blends, copolymers, network polymers and comb-branch polymers [12, 13]. Some modified forms of the polymer are fully amorphous and differ with the phase state of the semicrystalline PEO. Clarifying ion transfer in the fully amorphous nanocomposite polymer electrolytes will help us to understand the conductive mechanism and find a better way to enhance the ionic conductivity.
In this investigation, fully amorphous comb-branch polymers with different ether oxygen sidechain lengths were synthesized. A new type of nanocomposite polymer electrolyte was prepared with comb-branch polymers, LiCF3SO3 and SiO2 nanoparticles. The physical and electrochemical characteristics of ceramic nanoparticles in comb-branch polymer electrolytes with different sidechain lengths have rarely been reported. The amorphous samples have been characterized for their thermal and transport properties, and the effect of the filler on the conductivity is shown to be highly dependent on the sidechain length of the polymer matrix and the nanoparticle content; the maximum conductivity of 2.8×10−4 S cm−1 was obtained at 28 °C. A model is given to interpret the mobility of Li+ ions in a nanocomposite polymer electrolyte with variable SiO2 content.
Experimental
Materials
Poly(styrene-co-maleic anhydride) (PS) and poly(ethylene glycol) monomethyl ether (PEGME) with molecular weights of 350, 550 and 750 were purchased from Aldrich and used without further purification. Toulene-p-sulfonic acid (Shanghai First Chemical) was dried under vacuum for 12 h before use. Methyl ethyl ketone was refluxed in the presence of P2O5 for 4 h, then distilled. Methanol was dehydrated using 4 Å molecular sieve. LiCF3SO3 was purchased from Aldrich and dried under vacuum at 30 °C for 48 h before use. The SiO2 filler, ~7 nm particle size (Aldrich), was dried at 250 °C under vacuum for 24 h before use.
Synthesis of the comb-branch polymers
The reactions involved are outlined in Scheme 1 and the resulting comb-branch host polymers having different PEGME side chains are denoted by PS350, PS550 and PS750, depending upon the value of n (n=7, 12 and 17, respectively). More details of the synthesis are available in previous work [14].

Synthesis of the comb-branch polymers PS350, PS550 and PS750
Preparation of nanocomposite polymer electrolytes
The polymer was dissolved in acetone to obtain a polymer solution. Then the dry SiO2 nanoparticles with a calculated weight percent were firstly dispersed in the polymer solution in an ultrasonic cell for 4 h, and an appropriate amount of LiCF3SO3 (EO/Li=16/1) was added to the definitive volume of the polymer solution. After mixing thoroughly, the solution was cast on a home-built cell (see Scheme 2) [15]; the acetone was allowed to evaporate slowly at ambient temperature. The samples obtained were dried thoroughly under vacuum at 60 °C for 48 h.

A diagram of the cell for the preparation of solid polymer electrolyte thin films: 1, screw; 2, Teflon ring; 3, Teflon plate; 4, film
DSC and TGA measurements
DSC measurement of the polymer electrolytes containing different SiO2 contents (0–40 wt%) was carried out using Perkin-Elmer DSC-7 system; the heating rate was 10 K/min from −70 °C to 150 °C in a pure nitrogen atmosphere. TGA measurements were also performed on samples of about 2.5 mg using a Perkin-Elmer TGA-7 system in the temperature range from 0 °C to 500 °C under a flow of dry N2.
Conductivity measurements
Impedance measurements were carried out by Autolab/PG30 electrochemical analyzer system (ECO Chemie, Netherlands) in a frequency range from 0.1 Hz to 1 MHz at a perturbation signal of 5 mV. The polymer electrolyte film, sandwiched between two stainless steel electrodes in a sealed Teflon cell, was placed in a glass vessel equipped with a temperature-controlled heater. All the experiments were under a flow of dry N2; the experimental temperature range was from 20 to 90 °C. The impedance diagram is similar to that with added Al2O3 filler and the conductivity data were calculated by a.c. impedance according to our previous method [15].
Results and discussion
Figure 1 compares the TGA curves of the polymer electrolyte PS550–LiCF3SO3 (curve b) with the nanocomposite polymer electrolyte PS550–LiCF3SO3–10%SiO2 (curve a); no weight loss is observed and the films retain their thermal stability well up to 100 °C, indicating no solvent existed in the films. In the temperature range of 150–300 °C the films have 20 wt% loss, which may be caused by the side chains breaking away from the main chain. Above 300 °C the main chain of the comb-branch polymer begins to decompose on increasing the temperature. The decomposition temperature of curve a is 20 °C higher than that of curve b, which obviously shows that the dispersion of the ceramic nanoparticles can improve the thermal stability of the polymer electrolyte; in addition, the mechanical strength of the polymer electrolytes improves substantially. The improvement of these properties is important in view of their application for rechargeable lithium batteries.

TGA curves of (a) the nanocomposite polymer electrolyte PS550–LiCF3SO3–10%SiO2 and (b) the polymer electrolyte PS550–LiCF3SO3
Figure 2 shows the DSC measurement carried out on the polymer electrolyte PS550–LiCF3SO3. It can be seen that there are two glass transitions in the temperature range studied: one is at 40–70 °C, corresponding to the main-chain glass transition of PS550 (α-glass transition temperature, T α); the other is in the temperature range −50 to −40 °C (not accurately measured) and belongs to the glass transition of the side chain (β-glass transition temperature, T β), because T β is near the glass transition temperature of oligo-PEO. The comb-branch polymer exhibiting multiple glass transitions is consistent with the report of Lin et al. [16]. The T α values of PS550–LiCF3SO3–SiO2 with different nanoparticle contents were measured and are shown in Fig. 3, from which we can see that T α values of the nanocomposite polymer electrolytes increase with the addition of the nanoparticles, that is to say, the main chain of the host polymer becomes more and more rigid with an increase of SiO2 content [17]. Mertens et al. [18] reported that the chain flexibility in host polymer–Li salt composites has considerable influence on the migration of ions in the polymer matrix, since ion transport varies with the chain segmental motion of the amorphous polymer electrolyte. Increasing the volume fraction of the amorphous domains and decreasing the glass transition temperature appear to be the main rules to obtain better ionic conductivity.

DSC experimental curve for the polymer electrolyte PS550–LiCF3SO3

The glass transition temperature of the PS550 main chain as a function of SiO2 nanoparticle content
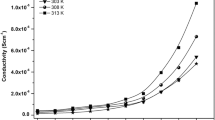
In Fig. 3 it can be concluded that the main chains become more and more rigid with an increase of SiO2 content; under this circumstance, it should lead to a drop in the ionic conductivity of the nanocomposite polymer electrolytes. However, the experimental results are contrary to this from investigation of the ionic conductivities of these nanocomposite polymer electrolytes, and all samples show a conductivity enhancement in comparison to the polymer electrolyte free of SiO2. Besides, the samples appear to be fully amorphous, as demonstrated by the absence of melting peaks observed in the temperature range of Fig. 2, so the conductivity enhancement cannot be attributed to the crystalline phase transition in the PEO–salt–filler since the comb-branch polymers are fully amorphous. The only reason for the conductivity enhancement is by the nanoparticles–polymer–salt interaction, especially the interaction of the nanoparticles’ surface with the side chain of the comb-branch polymers, but not with the backbone of the main chain which has little effect on the mobility of Li+ ions at ambient temperature.
Table 1 shows the conductivities of polymer electrolytes based on different side-chain lengths with the SiO2 addition. It is interesting to find that the system PS750–LiCF3SO3 has the largest conductivity change value among the polymer–salt systems studied, which indicates that the relatively longer side chain of the comb-branch polymer electrolytes easily produces higher ionic conductivity after the addition of the nanoparticles. The reason may be that PS750, with its longer side chain, is more facile to encapsulate and interact with the nanoparticles.
For this amorphous solid polymer electrolyte, the temperature dependence of the ionic conductivity generally follows the Vogel–Tammann–Fulcher (VTF) equation:
where A is a constant, E is the activation energy for ionic conduction and T 0 is the equilibrium state glass transition temperature. The activation energy E for ionic conduction below and above T α can be determined through the log σ versus 103(T−T 0)−1 plots, from the slope of which the value of E is obtained.
Figure 4 exhibits the VTF plots of the ceramic-free polymer electrolyte PS550–LiCF3SO3 and PS550–LiCF3SO3–5%SiO2 and PS550–LiCF3SO3–10%SiO2 nanocomposite polymer electrolytes. In these plots, two straight lines intersect at about the T α of the polymer electrolyte and exhibit special dual VTF behavior that is due to the α-transition temperature of the main chain [16]. The slope value above T α is lower than that below T α, which indicates that a lower activation energy and that the ion mobility above T α is more facile compared to that below T α. Consequently, it is concluded that the flexibility of the main chain in the nanocomposite polymer electrolyte has an influence on the mobility of ions in the polymer matrix at high temperatures (above T α).

Comparison of the VTF plots of the ceramic-free PS550–LiCF3SO3 polymer electrolyte, and the nanocomposite polymer electrolytes PS550–LiCF3SO3–5%SiO2 and PS550–LiCF3SO3–10%SiO2
Figure 5 shows a pictorial model of Li+ ion mobility in the solid polymer electrolyte under an applied voltage. Figure 5a simply depicts how Li+ ions move in the comb-branch polymer electrolyte; Fig. 5b and Fig. 5c show the reason for the conductivity enhancement with the addition of SiO2. Molecular dynamics simulations suggest that Li+ ions are complexed to the side chain of the polymer through approximately five ether oxygens [19]. The ionic conductivity is mainly attributed to the cation–polymer interactions and the motion of the comb-branch polymer side chain; the cation transport is described as the motion of Li+ species between complexation sites assisted by the segmental motion of the side chain. Local segmental motion of the side chain governs the long-range mobility of the charge carriers; the side chain is more important for conduction in the comb-branch polymer electrolyte than that of the main-chain backbone at ambient temperature. The addition of the SiO2 results in an increase of the conductivity by more than one order magnitude (see Table 1), which can be interpreted in terms of the acid–base approach to the mechanism of interaction in the polymer–salt–nanoparticles system. Here SiO2 can be considered as a weak Lewis- and/or Brønsted-type acid and may easily compete with the ethylene oxygen of the side chain for the formation of complexes with lithium cations. The SiO2 nanoparticles would act as cross-linking centers for the segments, lowering the polymer chain reorganization tendency and promoting an overall structure stiffness. This in turn may result in structural modifications occurring at the ceramic surface, which would provide conducting pathways for Li+ ions at the ceramic surface [7, 20]. The SiO2 nanoparticles also act by lowering ionic coupling, promoting salt dissociation via a sort of ion–ceramic complex formation, as well as it being demonstrated that the presence of the SiO2 fillers moves the Li+–CF3SO3 − reaction equilibrium toward the redissociation of ion pairs and an increase in the cation transport, thus accounting for the improvement in ionic transport. The results reported in this work confirm that in a nanocomposite polymer electrolyte the ceramic filler can perform both as a sort of “solid plasticizer” for the polymer chains by kinetically inhibiting their reorganization [21, 22], separating the polymer chains due to the filler which provides a large pathway for ions to diffuse at ambient temperature, and as a sort of a “solid solvent” by interacting with lithium salt ionic species [23, 24, 25, 26]; these interactions and Li+ ion mobility can be interpreted with the model in Fig. 5b. In our system the longer side chain easily interacts and encapsulates the nanoparticles, indicating that with the relatively longer side chain of the comb-branch polymer electrolyte it is easier to obtain higher ionic conductivity after the addition of the nanoparticles.

Pictorial model of the motion of Li+ ions in a solid polymer electrolyte: A ceramic-free polymer electrolyte; B nanocomposite polymer electrolyte containing a low SiO2 content; C a nanocomposite polymer electrolyte containing a high SiO2 content
Depending on the filler concentration, a maximum conductivity of 2.8×10−4 S cm−1 is obtained at 28 °C for the sample PS550–LiCF3SO3–10%SiO2. The conductivity as a function of SiO2 nanoparticle concentration is shown in Fig. 6. It should also be noted in this case that the conductivity is not a linear function of the filler concentration. At first, the conductivity increases with the addition of SiO2 and reaches a maximum value at 11 wt% SiO2; after that, the conductivity of the nanocomposite polymer electrolyte decreases gradually. This phenomenon is similar with the PEO–salt–filler system and can be explained in two ways: one is that the decrease in conductivity at higher filler content can be due to the higher stiffening of the polymer host, not only for the main chain but also for the side chain. The other explanation is that due to the large surface area of the filler, a more efficient ionic transport in the polymer nanoparticles grain boundaries is achieved when the content of ceramic nanoparticles is low; however, when the SiO2 content is higher, the nanoparticles easily aggregate, because many Li+ ions cannot cross the nanoparticle/nanoparticle interface (see Fig. 5c), so there is a loss of conductive pathways with large additions of ceramic fillers, the net result corresponding to the final drop of the conductivity.

The conductivity of PS550–LiCF3SO3–SiO2 as a function of SiO2 nanoparticle content at 18 °C
Conclusions
The results reported here demonstrates that a new type of nanocomposite polymer electrolyte based on a fully amorphous comb-branch polymer matrix has been prepared and characterized. The thermal and transport properties of the samples have been measured. From the results, ceramic SiO2 nanoparticles can improve the thermal stability and mechanical strength of the polymer electrolyte. Depending on the filler content, a maximum conductivity of 2.8×10−4 S cm−1 is obtained at 28 °C. Because the polymer matrix is fully amorphous, the reason for the conductivity enhancement is not due to the change of the phase state after SiO2 addition, but these locally induced structural modifications result in an increase of the fraction of free Li+ ions which can move fast throughout the conducting pathways at the nanoparticles’ extended surface, and reflect enhancements in the ionic conductivity. The relatively longer side chain of the comb-branch polymer electrolyte encourages higher ionic conductivity with the addition of SiO2 nanoparticles.
References
Fenton D, Parker JM, Wright PV (1973) Polymer 14:589
MacGlashan GS, Andreev YG, Bruce PG (1999) Nature 398:792
Andreev YG, Bruce PG (2000) Electrochim Acta 45:1417
Nishimoto A, Agehara K, Furuya N, Watanabe T, Watanabe M (1999) Macromolecules 32:1541
Patric J (2001) Polymer 42:8629
Xu W, Siow KS, Gao ZQ, Lee SY (1998) Chem Mater 10:1951
Croce F, Curini R, Martinelli A, Persi L, Ronci F, Scrosati B (1999) J Phys Chem B 103:10632
Jarascon J-M, Armand M (2001) Nature 414:359
Wieczorek W, Zalewska A, Raducha D, Florjańczyk Z, Stevens JR (1998) J Phys Chem B 102:352
Croce F, Appetecchi GB, Persi L, Scrosati B (1998) Nature 394:456
Qian XM, Gu NY, Cheng ZL, Yang XR, Wang EK, Dong SJ (2001) Electrochim Acta 46:1829
Best AS, Ferry A, MacFarlane DR, Forsyth M (1999) Solid State Ionics 126:269
Croce F, Gerace F, Dautzemberg G, Passerini S, Appetecchi GB, Scrosati B (1994) Electrochim Acta 39:2187
Qi L, Lin YQ, Jing XB, Wang FS (2001) Solid State Ionics 139:293
Qian XM, Gu NY, Cheng ZL, Yang XR, Wang EK, Dong SJ (2001) J Solid State Electrochem 6:8
Lin YQ, Qi L, Chen DL, Wang FS (1996) Solid State Ionics 90:307
Capiglia C, Mustarelli P, Quartarone E, Tomasi C, Magistris A (1999) Solid State Ionics 118:73
Mertens IJA, Wübberhorst M, Oosterbaan WD, Jenneskens LW, Turnhout JV (1999) Macromolecules 32:3314
Meyer WH (1998) Adv Mater 10:439
Croce F, Persi L, Scrosati B, Serraino-Fiory F, Plichta E, Hendrickson MA (2001) Electrochim Acta 46:2457
Best AS, Adebahr J, Jacobasson P, Macfarlane DR, Forsyth M (2001) Marcomolecules 34:4549
Wieczorek W, Raducha D, Zalewska A, Stevens JR (1998) J Phys Chem B 102:8725
Kumar B, Scanlon LG (1999) Solid State Ionics 124:239
Krawiec W, Scanlon LG Jr, Fellner JP, Vaia RA, Vasudevan S, Giannelis EP (1995) J Power Sources 54:310
Scrosati B, Croce F, Persi L (2000) J Electrochem Soc 147:1718
Macfarlane DR, Newman PJ, Nairn KM, Forsyth M (1998) Electrochim Acta 43:1333
Acknowledgements
This project was supported by the National Natural Science Foundation of China (no. 20075028).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhao, F., Wang, M., Qi, L. et al. Properties of a nanocomposite polymer electrolyte from an amorphous comb-branch polymer and nanoparticles. J Solid State Electrochem 8, 283–289 (2004). https://doi.org/10.1007/s10008-003-0449-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-003-0449-x