
Abstract
Electrochemical characteristics for the nanoporous carbon|Et4NBF4+acetonitrile interface have been studied by cyclic voltammetry and impedance spectroscopy methods. The influence of the electrolyte concentration and thickness of the nanoporous electrode material on the shape of the cyclic voltammetry and impedance curves has been established and the reasons for these phenomena are discussed. A value of zero charge potential, depending slightly on the structure and concentration of the electrolyte, the region of ideal polarizability and other characteristics have been established. The nanoporous nature of the carbon electrodes introduces a distribution of resistive and capacitive elements, giving rise to complicated electrochemical behaviour. Analysis of the complex plane plots shows that the nanoporous carbon|Et4NBF4+acetonitrile electrolyte interface can be simulated by an equivalent circuit, in which two parallel conduction paths in the solid and liquid phases are interconnected by the double-layer capacitance in parallel with the complex admittance of the hindered reaction of the charge transfer or of the partial charge transfer (i.e. adsorption stage limited) process. The values of the characteristic frequency depend on the electrolyte concentration and electrode potential, i.e. on the nature of the ions adsorbed at the surface of the nanoporous carbon electrode. The value of the solid state phase resistance established is independent of the thickness of the electrode material.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Electrochemical and electrical double-layer characteristics of carbon electrodes have been studied for a long time, but there are many problems which have not been solved at the moment [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15]. Electrical double-layer characteristics of various carbonaceous materials are very important as these parameters determine the electrical behaviour of the electrical double-layer capacitors (EDLCs), where the electrical charge is stored in the double layer (as the Gibbs energy of adsorption) and is based mainly on electrostatic interactions (so-called physical adsorption). The very important advantages of EDLCs are their reversibility and the comparatively low temperature coefficient [1]. However, a very important problem with EDLCs is their relatively low energy density compared with the rechargeable batteries. It is well known that the performance specifications of an electrochemical capacitor, e.g. in terms of the relations between achieved power densities and corresponding energy densities, depend on the equivalent series resistance (ESR) and on the internal distribution of the electrode resistance (IER) in the pore matrix of the electrode [1, 2, 3, 4, 5].
The capacitance of EDLCs depends mainly on the specific surface area of the carbon material used for preparation of the electrodes, as well as on the chemical composition and geometrical structure of the cations and anions adsorbed. Theoretically, the higher the surface area of the activated carbon, the higher the specific capacitance that should be expected. However, the practical situation is more complicated and usually the capacitance measured does not have a linear relationship with the specific surface area of the electrode material. There are two main reasons for this phenomenon: (1) the double-layer capacitance, C dl, varies with various types of activated carbon that are made from the different types of precursor (through different processes and subsequent treatments) [1, 2, 3, 4, 5, 6]; (2) nanopores with a small diameter may not be accessible to the electrolyte solution, simply because the electrolyte ions, especially large organic ions and ions with the solvation cell, are too big to enter into the nanopores.
In the literature there are very large differences between the values of the electrical double-layer capacitance of the carbonaceous materials (CMs), ranging from 3 μF cm−2 for the basal C(0001) plane of stress-annealed highly oriented pyrolytic graphite, to 70 μF cm−2 for the polished graphite edge plane [1, 2, 3, 16, 17]. The very low non-faradaic differential capacitance value for the C(0001) plane can be explained on the basis that a substantial fraction of the potential drop between the carbon electrode and the solution occurs in a space-charge layer. The various physical models developed so far [16, 17, 18, 19, 20, 21] cannot be used for interpretation of the experimental impedance data for the porous CMs because the surface roughness factor for nanoporous carbon (NPC) is not a very well established parameter. However, the potential drop in the thin surface layer of CMs is very important [1, 2, 16, 17, 18].
It should be noted that carbon electrodes show a frequency-dependent capacitance even through the capacitance should be independent of frequency. This abnormal frequency dependence is called a distributed characteristic or “frequency dispersion” of the electrical properties and it results mainly from two origins [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15]. (1) It appears non-locally when a dimension of a system under study (electrode thickness or pore length) is longer than the characteristic length (for example, diffusion length or a.c. penetration depth), which is a function of frequency, and this type of distributed characteristic exits even when all the system properties are homogeneous and space invariant. (2) The distributed characteristic is attributed to various heterogeneities: geometric inhomogeneity, such as the surface roughness or the distribution of pore size, as well as the crystallographic anisotropy and the surface disorder of a polycrystalline electrode. A circuit element with the distributed characteristics cannot be exactly expressed as a combination of a finite number of ideal circuit elements, except in certain limiting cases.
Theoretical background
Beginning essentially with the work of de Levie [10, 12, 13], a large number of various models have been developed [1, 2, 8, 9, 11, 14, 15, 16, 17] to describe theoretically the experimental behaviour of the carbon or porous carbon electrodes. A very important direction is the investigation of the influence of the pore geometry on the data for impedance spectroscopy [14, 15]. Some authors used simple modifications of the classical Randles–Frumkin–Melik–Gaikazyan equivalent circuit (RFMG) [22, 23, 24, 25, 26, 27] (i.e. so-called Ho model [28]), involving a constant phase element or Warburg diffusion impedance modified according to the boundary conditions [1, 8, 25, 26], as well as the branched transmission line equivalent circuits [1, 8, 27]. It should be mentioned that the Ho et al. model [28] does not describe the impedance of a porous electrode, but describes the influence of the thickness of an electrode on the diffusion with the reflective boundary condition in the frame of the RFMG mechanism [8]. Paasch et al. [11] developed a theory for a macroscopically homogeneous porous electrode, where three main processes are considered: (1) ionic conductivity in the pore electrolyte and electronic conductance in the electrode (solid) phase; (2) charging the double layer on the inside porous electrode surface|liquid interface; and (3) a charge transfer reaction (c.t.r.) on the inside electrode surface or a partial c.t.r. in our conditions for the nearly ideally polarizable electrode [29]. The Paasch et al. model [11] includes also a finite diffusion of the ions due to the small pore size with reflective boundary condition [8, 11]. The averaged polarization at the porous surface was described by the diffusion equation with the linear source term representing the c.t.r. According to this theory, the position (x)- and time (t)-dependent polarization, \( \tilde E(x,t) \) (averaged for the porous surface), has been given (without regard to an unknown constant) as:
where φ1 is a value of the potential in the solid phase (taken independent of x, φ1≠f(x)) [11]. The potential for the electrolyte in the pore, φ2, is determined by the electrolyte conductivity, double-layer formation parameters and c.t.r. characteristics of the interface (or by the partial charge transfer characteristics for an “ideally” polarizable electrode). Averaging this potential over the volume element, containing many pores, gives the position- and time-dependent potential (φ2(x,t)). In the case of low conductance for an electrode material, the resistivity per unit length of the solid phase, ρ1, is important. After averaging the potential over the volume element Adx (with the c.t.r. conductance dG ctr=gctrdx, where g ctr is the charge transfer conductance per unit length; A is the electrode flat-gross section surface area), the potential for the electrode matrix, φ1, also becomes position dependent, i.e. φ1=f(x). The theory of Rouśar et al. [9] at \( \rho _1^{ - 1} \nabla ^2 \phi _1 + \rho _2^{ - 1} \nabla ^2 \phi _2 = 0 \) (ρ2 is the resistivity per unit length of electrolyte in the porous material leads to the relation:
where C 1 is the double-layer capacitance per unit length averaged similarly to the potential (C 1=ASpCdl, where S p is the area of the pores per unit volume, on which an electrical double layer can be formed, and C dl is the “usual” double-layer capacitance per unit surface area of the electrode material) and r 1 is the resistance per unit length of the solid electrode matrix.
The electrochemical impedance of an interface is determined by the potential drop, E el, over an electrode [11]:
where \( \phi _{1,{\rm{a}}} (d,\omega ) \) is the solid-phase potential at the contact side (x=d); \( \phi _{2,{\rm{a}}} (0,\omega ) \) is the electrolyte potential at the electrolyte side (x=0); ω is the a.c. angular frequency; and j a is the amplitude of the current density (reported to unit geometrical area of the electrode).
Using the usual electrochemistry conditions [11], the electrode impedance is obtained as:
with:
where ω1 is a characteristic frequency, related to the finite “field diffusion”, and the characteristic frequency k and “field diffusion constant” K are defined as:
and:
In Eq. 7, j 0 is the exchange current density, R g and F are the universal gas and Faraday constants, respectively, and n is the number of electrons transferred in the elementary step of a redox reaction. According to Eqs. 8 and 7, the so-called “field diffusion constant”, K, depends only on the ohmic resistivities ρ1 and ρ2 and on the double-layer capacitance, and the ratio of the exchange current density to the double-layer capacitance determines the characteristic frequency k.
The a.c. decay length λ is defined as [11]:
If finite diffusion to the planar porous electrode takes place, then the value of the charge transfer resistance is defined as:
where \( {\rm{i}} = \sqrt { - 1} \;{\rm{and}}\;z(\omega ) = {1 \mathord{\left/ {\vphantom {1 {y(\omega )}}} \right. \kern-\nulldelimiterspace} {y(\omega )}} \) is the value of the volume-averaged hindrance impedance, describing the deviation of a system from the conditions of the real charge-transfer resistance. For a simple electrochemical charge-transfer reaction, the characteristic frequency ω2 is given as:
where D denotes the diffusion coefficient of the electroactive species and k het=k ox+k red is the rate constant. For the macroscopically homogeneous surfaces, Eq. 10 seems to explain the dominant dependence that only has to be corrected for the fact that the diffusion is finite owing to the small nanopore size.
The corrected hindrance impedance for a single kind of diffusing species inside the pores (i.e. on the inside electrode surface), when the boundary condition is not a transmissive one, will be of the form:
with:
where l p denotes the characteristic pore dimension [11].
Compared with the case of a simple c.t.r., in the Paasch et al. model I there are two additional parameters, ω2 and ω3, i.e. parameters characterizing the behaviour of the porous electrodes if finite diffusion inside the pores takes place [8].
Experimental
The electrodes comprised an aluminium foil current collector and the active material layer. The active material used consisted of nanoporous carbon (prepared from TiC starck grade C.A. by the chlorination method according to the published preparation scheme [2, 6, 30, 31, 32, 33], the mixture of the binder (polytetrafluoroethylene, 60% solution in H2O, Aldrich) and carbon black (Aldrich). Carbon black was added to decrease the ohmic resistance of the electroactive material. This mixture was laminated on the Ni foil and pressed together to form a very flexible layer of the active electrode material. The density of the nanoporous carbon electrode (NPCE) decreases with the increase in thickness of the NPCE, l NPCE (Fig. 1), but the BET surface area and other characteristics of the NPCE, obtained from N2-adsorption parameters [34], were practically independent of l NPCE. After drying and plating under vacuum, the NPCE was covered by a very pure Al layer on one side [2, 30, 31, 32, 33]. Then the Al-covered carbon layer was spot-welded in an Ar atmosphere to the Al foil current collector. The limits of ideal polarizability of the Al foil, established by cyclic voltammetry as well as by impedance spectroscopy methods, are wider than those for the NPCE studied.

Dependence of NPCE material density ρ on the thickness l of the electrode
The electrolytes used were prepared from very pure acetonitrile (AN) (0.003% H2O, Riedel-de Haën), stored over the molecular sieves before use, and from very dry Et4NBF4 salt (99.9%, 0.005% H2O; Pred Materials International), additionally purified and dried [18, 30, 31, 32, 33]. A three-electrode standard glass cell with a very large counter electrode (apparent area ~30 cm2), prepared from carbon cloth, was used. In some experiments, a very large Pt-mesh counter electrode was used, but the experimental results (i.e. the values of current density measured) were independent of the counter electrode material used. The reference electrode was an aqueous saturated calomel electrode (SCE in H2O) connected through an electrolytic salt bridge (0.1 M LiClO4 in H2O|0.1 M Et4NBF4 in AN) to the measurement system [18]. Very pure Ar (99.9999%, AGA) was used for saturating the solutions. Specific surface area, pore size distribution, micropore volume, micropore area and other parameters were measured using the Gemini 2375 (Micromeritics) system and calculated according to the methods described [34]. Some more important characteristics obtained are given in Table 1 and in Fig. 2. According to the data in Fig. 2, the nanopores with a pore diameter d=1.1–1.2 nm prevail on the surface of nanoporous carbon. The specific area of nanoporous carbon ID 1727, obtained by the Brunauer, Emmet, and Teller (BET) method, has a surface area of 1100±200 m2 g−1, practically independent of l NPCE. Comparison of these data with those published [2, 6] indicates that the specific surface area for ID 1727 is somewhat higher than that for nanoporous carbon ID 711 but somewhat lower than that for ID 1369 [6].

Pore size distribution for nanoporous carbon ID1727
The results of AFM studies for nanoporous carbon ID 1727 show that the NPCE has a very rough and complicated surface structure.
Results and discussion
Cyclic voltammetry curves
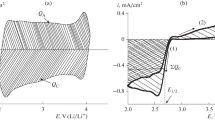
The cyclic voltammetry (j vs. E) curves for 0.1 M and 1.0 M Et4NBF4+AN solutions, obtained at low scan rates of potential v=dE/dt≤10 mV s−1 (Fig. 3a), have nearly mirror-image symmetry of the current responses about the zero current line [j=current density, obtained using the flat cross-section (geometrical) surface area of the NPCE]. Accordingly, the NPCE is ideally polarizable in the region of potentials from −1.4 to 1.4 V (vs. SCE in H2O) in the Et4NBF4+AN solutions. The experimental data show that the shape of the j vs. E curves is independent of the number of current cycle, n, if n≥3. Thus, the NPCE demonstrates stable electrochemical properties in this region of potentials, i.e. in the region of ideal polarizability. At a fixed electrolyte concentration, c el, the current responses increase almost in the expected way with large ν values (Fig. 3a), but with increasing v≥20 mV s−1 the cyclic voltammograms become distorted from the mirror-image symmetry [1, 2]. The deviation of the j vs. E curves from the mirror-image symmetry is more pronounced for the less concentrated electrolyte solutions, in good agreement with published data [1, 2, 3, 4, 5, 6].

(a) Current density vs. potential (j vs. E) curves for l NPCE=60 μm thick NPCE in 0.1 M Et4NBF4+AN at different potential scan rates v (mV s−1), noted in the figure; (b) C vs. E curves for NPCE ID1727 in 0.1 M Et4NBF4+AN interface obtained from j vs. E curves at different scan rates: (1) −0.1, (2) −0.2, (3) −0.5 and (4) −1 mV s−1; (c) E min vs. potential scan rate curves for NPCE ID1727 in 0.1 M Et4NBF4+AN at different l NPCE values (noted in figure), obtained from j vs. E curves for positive directions of the potential scan
If the series resistance R s≠0 and I≠0, then for cyclic voltammetry the applied voltage E t is given as:
where E 0 is the initial potential, t is time and E 2 is the upper limit of the potential sweep. Correspondingly, the voltage applied during the negative direction of the potential sweep is:
Assuming that R s=constant and I=f(t), the following expressions for the positive current:
and for the negative current:
can be obtained when C is the series capacitance of the electrode. However, according to the experimental results, this effect depends noticeably on l NPCE and this effect is very large for the thicker NPCE in less concentrated Et4NBF4 solutions. Thus, at higher scan rates for thicker electrodes there are very well expressed distortion effects in the j vs. E curves, caused by the significant resistivity of the electrolyte in the porous material, as well as probably by the resistivity of the porous electrode material (i.e. by the significant series resistance of the experimental system). The first effect has been explained by the onset of the so-called “electrolyte starvation” [1, 35], which is associated with the withdrawal of the electrolyte ions from the pore bulk electrolyte due to adsorption of the ions on the double-layer interphases when they become charged. Comparison of the j vs. E curves for different electrolyte concentrations shows that the scan rate at which the deviation of the j vs. E curves from the ideal behaviour starts decreases with the dilution of the electrolyte solution. Thus, the solution resistance is important but, on the other hand, the establishment of the adsorption equilibrium in the nanopores is a very slow process, which is caused by the very small “effective” diffusion coefficient values of the ions in the nanopores influenced by the effective diffuse layer thickness at the electrode surface, as well as by the increase of the effective Debye screening length with the dilution of the electrolyte [1, 2, 6, 18, 33, 36, 37].
According to the experimental results (Fig. 3a), in the j vs. E curves obtained at low scan rates, there is a minimum of current density in the potential region −0.1≤E≤0.5 V (vs. SCE in H2O), which, in the case of metal electrodes, is explained by the diffuse nature of the electric double layer in the region of total (or free) zero charge [18].
Noticeably better expressed current (i.e. capacitance) minima have been established for the 0.01 M and 0.005 M Et4NBF4+AN solutions (Fig. 3b), in good agreement with published results [2, 6]. The current density at ν=constant and E=E min=0.27 V vs. SCE decreases with dilution of the electrolyte. The potential of this minimum is practically independent of the direction of the potential scan if v≤2 mV s−1, as well as of the electrolyte concentration if c el≤0.1 M. Thus, to a first approximation, this minimum is probably caused by the zero charge potential (or by the total zero charge potential) of the nanoporous carbon electrode in AN [1, 2, 6, 15, 16, 17, 18]. The value of E min is practically independent of ν for the electrodes with l NPCE=60 μm and 100 μm (Fig. 3c), but for thicker electrodes there is a large dependence of E min on ν at higher scan rates, which is caused by the very slow desorption process of the Et4N+ cations from the negatively charged surface as well as by the “distortion” effect [1, 2, 6, 35]. For the 1.0 M Et4NBF4+AN system (l NPCE<150 μm) the dependence of E min on ν as well as on the potential scan direction is very small, explained by the smaller effective Debye length values for the electrolyte ions, i.e. by the quicker establishment of the adsorption equilibrium [1, 2, 6, 36].
Differential capacitance vs. potential curves
The values of the differential capacitance, corresponding mainly to the double-layer capacitance, can be obtained as:
if we assume that the capacitance C=constant and if the system series resistance R s→0 or if the current density j→0. It is to be noted that, in real conditions, Eqs. 14, 15, 16, 17, 18 can be used for obtaining the capacitance values only in the region of low potential scan rates if the values of the current are very small, as the potential drop (IR drop) losses are negligible only at these conditions and the current response is essentially of a pure capacitor [1, 2, 5, 6, 35].
The C vs. E curves in Fig. 3b, obtained from j vs. E curves, show that at E≈0.27±0.01 V (vs. SCE) the very well expressed capacitance minimum (with the potential of the minimum, E min) in the C vs. E curves has been observed for c≤0.1 M solutions. According to the experimental data, for 0.1 M Et4NBF4+AN solutions in the limited region of the electrode potential, corresponding to the zero charge potential E σ=0, the capacitance only weakly increases with the decrease of v 1/2. The calculated values of C min (F cm−2) (Table 2) increase with l NPCE (i.e. with the volume of the effectively working material). For that reason, the values of C have been recalculated into units of F g−1, where the active weight of the nanoporous carbon electrode has been taken into account. According to the results of the calculations, the value of C (F g−1) obtained is practically independent of l NPCE if l NPCE≤150 μm. The somewhat lower values of C for thicker electrodes can be explained by the noticeable diffusion limitations in the nanoporous electrode matrix. The lower values of C for 0.1 M Et4NBF4+AN solution compared with the 1.0 M solution can be explained by the diffuse nature of the electrical double layer at E≈E min. It should be noted that the values of C established from the j vs. E curves at ν≤5 mV s−1 are in good agreement with the C s vs. E curves [where C s is the series capacitance, calculated from the complex plane (Z″ vs. Z′) plots] measured at an a.c. frequency f≤10 mHz.
According to the experimental data (Fig. 4a, b), the series differential capacitance for the NPCE with thickness l NPCE=60 μm only very slightly depends on frequency in 1.0 M Et4NBF4 at f≤0.1 Hz, and for the 0.1 M electrolyte at f≤5×10−2 Hz. The NPCE with l NPCE=250 μm demonstrates the almost equilibrium capacitance values only in the 1.0 M solution at f≤5×10−3 Hz (Fig. 4a, b). Thus the frequency value at which the adsorption equilibrium has been established (f eq) depends on l NPCE and c el (i.e. on the Debye screening length) and f eq decreases with increasing l NPCE. At higher frequency, C s depends significantly on f, which is caused by the very small values of a.c. penetration depth (Eq. 9) compared with the pore length. At |E|>E min, there is a noticeable dependence of C s as well as of f eq on E, indicating that the effective Debye screening length plays an important role in establishment of the adsorption equilibrium. The dependence of the C s vs. f plot characteristics on the electrode potential (Fig. 5a, b) demonstrates that the limiting Gibbs adsorption is higher for BF4 − anions than for Et4N+ cations [37].

C s vs. E curves for NPCE ID1727 in (a) 0.1 M Et4NBF4+AN and in (b) 1 M Et4NBF4+AN at different l NPCE values, obtained from Z″ vs. Z′ curves at an a.c. frequency of 0.005 Hz

C s vs. f curves for NPCE ID1727 in (a) 0.1 M Et4NBF4+AN and (b) in 1 M Et4NBF4+AN at l NPCE=100 μm and different potentials (V vs. SCE), noted in the figure
Analysis of impedance data
The Z″ vs. Z′ plots [so-called Nyquist plots (Figs. 6 and 7a)] were measured for the nanoporous carbon|xM Et4NBF4+AN solution interface in the range of a.c. frequencies from 1×10−3 to 1×105 Hz and potentials from −1.4 to 1.4 V (vs. SCE in H2O) at fixed E=constant. The Z″ vs. Z′ plots have been simulated according to the modified Frumkin–Melik–Gaikazyan–Randles model (where formally the Warburg-like diffusion impedance takes into account the finite diffusion due to the small pore size with reflective boundary conditions [8]) (circuit I in Fig. 7b), the Paasch et al. model [11] (circuit II) or the modified Meyers et al. model [6, 36, 37, 38, 39]. In the Meyers et al. model [38], the charge transfer and double-layer charging at the surface, the intercalation of ions (H+ from H2O reduction) and solid phase diffusion inside the nanoparticle, as well as the effect of an insulating film at the nanoparticle surface, have been taken into account [38]. The better fit shown in Figs. 6 (a and b) and 7a (solid lines) has been established for the Paasch et al. model I [11, 39], in which two parallel conduction paths in the solid and liquid phase are interconnected by the double-layer capacitance in parallel with the complex admittance of the hindered reaction of the charge transfer process inside the electrode surface. The experimental results obtained demonstrate that the shape of the Z″ vs. Z′ plots depends noticeably on E (Fig. 6a) as well as somewhat on c el (Fig. 6b) and l NPCE (Fig. 7a). The complex plane plot for the Et4NBF4+AN electrolyte consists mainly of two parts: the depressed semicircle at higher a.c. frequencies (f≥40 Hz) with the characteristic frequency f max (given in Table 2), and the double-layer capacitance region at lower frequencies (so-called “planar electrode” section, where the finite length effects prevail). The shape of the Z″ vs. Z′ plots indicates that there are two limiting processes (the slow diffusion and slow heterogeneous adsorption steps [37, 38, 39, 40, 41], with the faradaic charge transfer or partial charge transfer process) affecting the rate of the adsorption process of the electrolyte ions at the nanoporous carbon|acetonitrile interface at f>40 Hz [1, 2, 22, 23, 24, 28, 36, 37, 38, 39, 40, 41]. Differently from the nanoporous carbon ID 711|AN+0.1 M Et4NBF4 interface [2] and from the ID 1369|0.1 M LiClO4 or 0.1 M LiBF4+AN systems [6], there is no very well expressed so-called nanoporous region in the Z″ vs. Z′ plots [i.e. there is no section in the Z″ vs. Z′ plot where the slope α′=45°, characteristic of the diffusion-limited process in the nanopores [1, 2, 6, 34, 36, 38, 39, 40, 41], except at E min (Fig. 6a)]. Accordingly, the “pore resistance” depends on the effective screening length of the ions in the electrical double layer [36, 38, 39, 40, 41, 42] and only in the region of E min for the 0.1 M Et4NBF4|NPCE interface there is a very compressed region, where the Z″ vs. Z′ plots are almost linear with the slope α′≈45°. It should be noted that the effective diffuse layer thickness (i.e. the inverse Debye length of the electrolyte ions) as a function of the electrode rational potential E=E x −E σ=0 is given as:

Complex plane (Z″ vs. Z′) plots for NPCE ID1727 (l NPCE=100 μm)|AN+1 M (a) and 0.1 M (b) Et4NBF4 interfaces at different electrode potentials, noted in the figure (symbols, experimental data; solid lines, simulation according to Paasch et al. model [11])

(a) Complex plane (Z″ vs. Z′) plots for NPCE ID1727|AN+1 M Et4NBF4 interface at potential E=0.2 V vs. SCE and different l NPCE values, noted in the figure (symbols, experimental data; solid lines, simulation according to Paasch et al. model [11]). (b) Equivalent circuits used for fitting the experimental complex plane plots: I, combined Frumkin–Melik–Gaikazyan and Randles (so-called Ho [28]) circuit, where R 1 is the high-frequency series resistance [R el=Z′(ω→∞)], CPE is the constant phase element, R ad is the charge transfer resistance, Z W is the Warburg-like diffusion impedance and C ad is adsorption capacitance; II, Paasch et al. circuit, where dY is the complex admittance of the hindered charge transfer reaction [involving the hindrance factor y(ω) of the charge transfer reaction with the charge transfer conductance g ct], dC dl is the double layer capacitance, dR 1 and dR 2 are the ohmic resistances of the porous electrode material and of the electrolyte in the pore, respectively, in the volume element Adx [11]
where κ is the Gouy length. The Debye length (equal to κ−1) is obtained as:
where \( \beta = \left( {k_{\rm{B}} T} \right)^{ - 1} \) (T is absolute temperature and k B is the Boltzmann constant; n is the bulk electrolyte concentration; ε is the dielectric constant of the solvent; and e is the elementary charge) [36, 42]. For that reason, it is impossible to obtain correctly the electrochemical parameters of the pores prevailing at the surface of the nanoporous carbon material, according to the published model [12, 13].
The dependence of the slope of the Z″ vs. Z′ plot on the electrode potential in the so-called planar electrode section (f<20 Hz) is caused mainly by the decrease of the effective screening length of the electrolyte ions with the increase of the surface charge density at the electrode|electrolyte interface [36, 42, 43]. According to the experimental data for Et4NBF4 in various solvents, the Walden product \( \left( {\eta ^0 \lambda _{{\rm{Et}}_{\rm{4}} {\rm{N}}^ + }^0 } \right) \) is practically independent of the solvent studied, and, therefore, the effective radius of Et4N+ ion in AN solution, to a first approximation, can be taken equal to the crystallographic radius of the cation (r CR= 0.343±0.002 nm) [44, 45] (η0 is the coefficient of viscosity for the pure solvent and λ0 is the limiting molar conductivity of AN with addition of Et4NBF4). Thus, the dependence of the shape of the Z″ vs. Z′ plot on E indicates that the effective pore dimension is of the same order of magnitude as the Debye length for the 0.1 M electrolyte+AN system (κ−1=0.66 nm for 0.1 M electrolyte in AN at T=298 K and E=E min).
The Z″ vs. Z′ plots (at f= constant) were used for the calculation of the values of the differential series capacitance C s (series circuit) and parallel capacitance C p (parallel circuit) (Fig. 8), as well as of the series resistance R s and parallel resistance R p values (ZView for Windows version 2.2, Scribner Inc.). According to the data obtained, the values of C s and C p for the NPCE with l NPCE=60 μm level off to the limiting value of C s(ω→0) at low frequency, f≤0.1 Hz, indicating that the diffusion-like step is a slow and limiting stage of the adsorption process of the Et4N+ ions in the region of E min as well as at E<<E min. With the increase of thickness of the electrode material the inflection frequency noticeably decreases, and for NPCE with l NPCE=250 μm in 0.1 M Et4NBF4 electrolyte, C s and C p level off only at f≤0.005 Hz. For 1.0 M electrolyte the level of C p and C s has been established at somewhat higher frequencies, f≥10−2 Hz. For a thinner NPCE there is no dependence of the inflection frequency on l NPCE. Comparison of the C s values obtained from impedance data with those from j vs. E curves demonstrates a good agreement between the differential capacitance values for both series at E≤E min if f≤10−2 Hz and v≤1 mV s−1.

C s (1, 2) and C p (3, 4) vs. frequency dependences for NPCE ID 1727 in 0.1 M (a) and 1 M (b) Et4NBF4+AN solutions at the potentials (vs. SCE) 0.2 V (1, 3) and −1.4 V (2, 4) for different l NPCE values: 60 μm (1, 3) and 250 μm (2, 4)
The results in Fig. 9a are in good agreement with these conclusions, as the values of the phase angle δ≈−90° for the NPCE with l NPCE=60 μm in 1.0 M electrolyte at E≤−1.4 V indicate mainly the adsorption stage limited process of ions on the NPCE at f≤0.01 Hz. The values of |δ|≤90° for the thicker NPCE (l NPCE>150 μm) and less concentrated electrolytes (Fig. 9a, b) indicate the mixed kinetics (slow diffusion followed by the slow adsorption step) of the adsorption of ions at the NPCE. It should be mentioned that for the NPCE with l NPCE=250 μm|0.1 M electrolyte interface the adsorption equilibrium has been established at f≤0.005 Hz. The dependence of |δ| on the electrode potential at f=constant indicates that the establishment of the adsorption equilibrium depends on the effective charge of the adsorbed ions (mixed kinetics for anions) (Fig. 9a, c).

(a) Phase angle vs. frequency dependences for NPCE ID1727 (l NPCE=60 μm) in 1 M Et4NBF4+AN solution at different potentials (V/SCE, noted in the figure). Phase angle vs. frequency dependences for NPCE ID1727 in 1 M (b) and 0.1 M (c) Et4NBF4+AN solution at E=1.4 V (vs. SCE) for different l NPCE, noted in the figure
The shape of the Bode amplitude (|Z| vs. logω) plots for various systems, obtained at different electrode potentials with E=constant, depends on c el as well as on l NPCE. The higher values of |Z| for the 0.1 M Et4NBF4 solution are caused mainly by the higher values of the pore resistance (Fig. 6a, b). At 1×10−1≤f≤1×103 Hz, there is noticeable dependence of |Z| on frequency at |E|≠E min, and the higher values of |Z| for NPCE at E σ=0 are mainly caused by the higher values of R pore.
Analysis of complex plane plots
The complex plane plots, measured and simulated for the NPCE|xM Et4NBF4+AN interface at different electrode potentials (Figs. 6, 7 and 10), can be divided into four main sections. At high a.c. penetrability [at very low a.c. frequency, f≤1×10−2 Hz, and thus at very high values of λ (Eq. 9)] the NPCE behaves like a planar electrode since the a.c. penetration depth λ is large and the a.c. signal detects a very large amount of the pore volume. However, according to the data in Figs. 6a and 10, in this region of f, the NPCE|AN interface demonstrates the non-ideal constant phase element (CPE) behaviour and constant phase exponent values α of less than 1.0 have been established (Table 2). The values of α depend on the electrode potential and are lower at |E|>E min, indicating that the specific adsorption of various ions at different electrode potentials (cations at E<E min and anions at E>E min) is possible [2, 6, 36]. On the other hand, at c el= constant, the dependence of α on |E| is weak and this result indicates that the effective working surface area of the electrode only very weakly depends on the Debye screening length at f≤0.01 Hz [2, 6, 36, 39, 42, 43].

Complex plane (Z″ vs. Z′) plots (a, b) for ID1727 (l NPCE=250 μm)|AN+1 M Et4NBF4 interface at different electrode potentials, noted in the figure (symbols, experimental data; solid lines, simulations) according to (a) the modified Randles–Frumkin–Melik–Gaikazyan model [8, 11, 22, 23, 24, 41] and (b) the Paasch et al. model I [11]
However, according to the model [43], the deviation of α from unity is caused mainly by the pore size distribution in the nanoporous material studied. It should be noted that more detailed computer simulations are inevitable, but the somewhat higher deviation of α from unity for ID 1727, compared with ID 711 and ID 1369 [2, 6], indicates that the nanoporous carbon ID 711 has a narrower pore size distribution, which is in good agreement with the BET data (Table 1 and Figs. 1 and 2 and the corresponding published data [2, 6]). The lower values of α at E>E min than at E≤E min indicate that the “effective” radius of the anions is smaller than the “effective “ radius of the cations [44, 45].
At lower penetrability (at higher frequency), the penetration depth is smaller than the length of the pores, so that the a.c. signal detects only a part of the pore volume. This is called the porous section of the Z″ vs. Z′ plots, and the phase angle approaches −45° in this region of a.c. frequency (Fig. 6). In addition, there is a transition section between the porous and the planar sections in the Z″ vs. Z′ plots at E=E σ=0.
At higher frequencies, there is a very well expressed, slightly depressed, semicircle in the Z″ vs. Z′ plot, with the depression angle, β, decreasing with the negative polarization of the electrode and with dilution of the electrolyte. It should be noted that the value of β equal to zero corresponds to the purely charge transfer limited heterogeneous process [β=45° corresponds to the diffusion limited stage, and 0<β<45° (depressed semicircle) corresponds to a charge transfer process at the inhomogeneous interfaces] [2, 6, 37, 38, 39, 40, 41, 42]. The value of β≤10° for the 1.0 M Et4NBF4+AN|NPCE interface (Fig. 6b) indicates that the fast heterogeneous adsorption step (i.e. the partial charge transfer reaction) is probably the rate-determining process for this system at f≥40 Hz. For the less concentrated Et4NBF4+AN solution, the kinetically mixed (diffusion and heterogeneous adsorption limited) process has been established.
Results of non-linear regression analysis (ZView for Windows version 2.2, Scribner Inc.) for the Z″ vs. Z′ plots show (Fig. 10a) that, to a first very rough approximation, these plots can be formally simulated using the modified Randles–Ershler-like equivalent circuit with the chi-square function χ2≥1.8×10−3 and weighted sum of squares Δ2≥0.25 for the 1.0 M Et4NBF4 solution (Fig. 7b, scheme I), where the generalized finite length Warburg element (GFW) with the reflective boundary condition (finite length transmission line terminated with an open circuit) is expressed as [46] (ZView for Windows version 2.2, Scribner Inc.):
where R D is the so-called limiting diffusion resistance, the frequency parameter T is expressed as T=L 2/D, where L is the effective diffusion layer thickness and D is the effective diffusion coefficient of a particle; αW (Fig. 11a, b) is a fractional exponent varying from 0 to 1. The values of αW higher than 0.5 indicate the deviation of the NPCE|AN+xM Et4NBF4 system from the classical semi-infinite diffusion model [22, 23, 24], increasing with dilution of the electrolyte and with the decrease of the active NPCE layer thickness and negative polarization of the NPCE. The parallel charge transfer (or adsorption) resistance, R ct, as well as diffusion resistance, R D (Fig. 11a, b), increase with increasing the negative electrode potential, and with decreasing the electrolyte concentration and thickness of the NPCE active layer. The frequency parameter T has minimal values in the region of E σ=0 and T increases with l NPCE and c el.

(a) Dependence of R D, T and αw on E( l NPCE=100 μm) and (b) on the active material layer thickness for 1.0 M (filled symbols) and 0.1 M (open symbols) Et4NBF4+AN electrolyte (symbols correspond to the following parameters: squares, R D; triangles, T; circles, αw)
However, a better agreement between simulations and experimental data for the NPCE|xM Et4NBF4+AN interface with the χ2 function (χ2≤6×10−4) has been established if the Paasch et al. model I [11] (circuit II in Fig. 7b) is used.
According to the results of fitting, the dependence of the characteristic frequency k ≡g ct/C 1=j0nF/CdlRgT on the thickness of the electrode material (Fig. 12) is mainly caused by the decrease of the charge transfer conductance per unit length with increasing l NPCE. The noticeably higher values of k for 1.0 M Et4NBF4 than for the 0.1 M solution are in good agreement with this conclusion and can be explained by the noticeable dependence of charge transfer conductance per unit length, g ct, on the electrolyte concentration (i.e. g ct increases with c el).

The dependences of the parameter k (calculated according to Paasch et al. model I) on the electrode potential for NPCE ID1727 in (a) 0.1 M and (b) 1 M Et4NBF4+AN solutions at different l NPCE, noted in the figure
The weak dependence of the characteristic frequency ω1 related to the finite “field diffusion”:
on l NPCE (Fig. 13a, b) can be explained by the dependence of ρ2 on l NPCE as well as of ρ2 and C dl on c el. The dependence of the characteristic frequency ω2=k het 2/D on l NPCE (Fig. 14) can probably be explained by the fact that the value of the diffusion coefficient D is an effective value and the increase of ω2 with l NPCE can be explained with the decrease of D in Eq. 11 (if we assume that k het≡k ox+k red is independent of l NPCE). The noticeably smaller values of ω2 for more concentrated electrolyte solutions can be explained by the higher effective diffusion coefficient values caused by the smaller effective Debye screening length for more concentrated solutions [2, 6, 39, 42, 43]. The fact that ω2 vs. E dependences have a maximum at E=E min clearly indicates that at these potentials the effective Debye screening length is maximal and, accordingly, the effective D values have to be minimal.

The dependences of the parameter ω1 (calculated according to Paasch et al. model I) on the electrode potential for NPCE ID1727 in (a) 0.1 M and (b) 1 M Et4NBF4+AN solutions at different l NPCE, noted in the figure

The dependences of the parameter ω2 (calculated according to Paasch et al. model I) on the electrode potential for NPCE ID1727 in (a) 0.1 M and (b) 1 M Et4NBF4+AN solutions at different l NPCE, noted in the figure
Figure 15 demonstrates that the value of the characteristic frequency ω3 decreases with increasing l NPCE (Eq. 12). The very small increase of ω3 with the decrease of l NPCE can be explained by the weak dependence of the effective value of D on l NPCE, as the characteristic nanopore dimension l p seems to be independent of l NPCE. The very small increase of ω3 with c el can probably be explained by the weak rise of D with the decrease of the effective Debye screening length [1, 2, 6, 36, 42]. Thus, very weak dependence of ω3 for NPCE on c el, E and l NPCE in comparison with ω0 and ω1 probably shows that the values of the effective diffusion coefficient D for ions in the nanopores depend very weakly on the so-called mass transport characteristics in the macropores, i.e. on the effective Debye screening length and solution molar conductivity values in the solution phase. Thus, the two parallel conduction paths in the solid and liquid phases are interconnected by the double-layer capacitance in parallel with the complex admittance of the hindered charge transfer reaction (the equivalent circuit in the inset of Fig. 7b).

The dependences of the parameter ω3 (calculated according to Paasch et al. model I) on the electrode potential for NPCE ID1727 in (a) 0.1 M and (b) 1 M Et4NBF4+AN solutions at different l NPCE, noted in the figure
Thus, according to simulations, the values of ω2, ω3<<ω0, and thereby the relative magnitudes of ω1, ω2 and ω3 determine the low-frequency behaviour of the Z″ vs. Z′ plots [11]. For that reason, at very low frequency, f≤5×10−2 Hz, there is a transition to a pure capacitive behaviour, since both the field diffusion and the species diffusion are finite. In the region of frequencies 0.2<f<10 Hz, the absolute value of the slope equal to π/4 at E=E σ=0 for the Z″ vs. Z′ plot was observed around ω2, where the diffusion of the species dominates in the solution phase. At higher frequency (f ≥40 Hz), the shape of the impedance spectrum is determined mainly by the ratio of the ω0 and ω1 values, i.e. without regard to the diffusion inside the pores [11].
The dependence of the function y on E as well as on c el and l NPCE indicates that the value of ρ2 (the electrolyte resistance per unit length inside a porous material) depends very noticeably on the electrolyte concentration as well as on the electrode polarization. The obtained value for ρ1 (resistivity of the electrode material per unit length) is independent of the electrolyte concentration studied.
Conclusions
The results of the impedance studies for the nanoporous carbon|acetonitrile+0.1 M and 1.0 M electrolytes (NPCE|Et4NBF4+AN) interface indicate that the series capacitance, series resistance, phase angle and other parameters depend noticeably on the concentration of the electrolyte, on the thickness of active material (nanoporous carbon layer) as well as on the electrode potential. In the region of electrode potentials 0<E<0.4 V (vs. SCE in H2O), a very well expressed differential capacitance minimum in the series capacitance vs. potential curves has been observed with the potential [2, 6] of the capacitance minimum, E min, equal to the zero (free) charge potential. The values of the differential series capacitance C s, obtained from the complex plane (Z″ vs. Z′) plots, depend noticeably on the a.c. current frequency if f≥1×10−2 Hz. Only at very small a.c. current frequency is C s practically independent of f, indicating that the nearly limiting capacitance values have been established. Non-linear regression analysis of the complex plane plots shows that the nanoporous carbon|acetonitrile+xM Et4NBF4 electrolyte interface can be simulated by the equivalent circuit, in which two parallel conduction parts in the solid and liquid phases are interconnected by the double-layer capacitance in parallel with the complex admittance of the hindered reaction of the charge transfer (or partial charge transfer in our case) process, i.e. by the Paasch et al. model I [11]. The values of the characteristic frequency depend on the electrolyte composition and on the electrode potential, i.e. on the nature and structure of the ions adsorbed at the surface of the nanoporous carbon electrode, as well as on the effective Debye screening length.
References
Conway BE (1999) Electrochemical supercapacitors: scientific fundamentals and technological applications. Kluwer/Plenum, New York, p 1
Lust E, Nurk G, Jänes A, Arulepp M, Nigu P, Permann L, Möller P (2002) J Condensed Matter Phys 5:307
Salitra G, Soffer A, Eliad L, Cohen Y, Aurbach D (2000) J Electrochem Soc 146:2486
Qu D, Shi H (1998) J Power Sources 74:99
Pell WG, Conway BE (2001) J Electroanal Chem 500:121
Lust E, Nurk G, Jänes A, Arulepp M, Nigu P, Möller P, Kallip S, Sammelselg V (2003) J Solid State Electrochem 7:91
Christen Th, Carlen MW (2000) J Power Sources 91:210
Macdonald JR, Johnson WB (1987) Fundamentals of impedance spectroscopy. In: Macdonald JR (ed) Impedance spectroscopy. Wiley, New York, pp 1–26
Roušar I, Micka K, Kimla A (1986) Electrochemical engineering, vol 2. Academia, Prague, pp 1–337
de Levie R (1990) J Electroanal Chem 281:1
Paasch G, Micka K, Gersdorf P (1993) Electrochim Acta 38:2653
de Levie R (1963) Electrochim Acta 8:751
de Levie R (1964) Electrochim Acta 9:1231
Keiser H, Beccu KD, Gutjahr MA (1976) Electrochim Acta 21:539
Keddam M, Rakomoto C, Takenouti H (1984) J Appl Electrochem 14:437
Randin JB, Yeager EB (1972) J Electroanal Chem 38:257
Gerisher H, McIntyre R, Shearson D, Storck W (1987) J Phys Chem 91:1930
Trasatti S, Lust E (1999) The potential of zero charge. In: White RE, Conway BE, Bockris JO’M (eds) Modern aspects of electrochemistry, vol 33. Kluwer/Plenum, New York, pp 1–216
Amokrane S, Badiali JP (1989) J Electroanal Chem 266:21
Amokrane S, Badiali JP (1991) Analysis of the capacitance of the metal-solution interface. In: Bockris J O’M, Conway BE, White RE (eds) Modern aspects of electrochemistry, vol 22. Plenum, New York, pp 1–95
Lust E, Jänes A, Lust K, Väärtnõu M (1997) Electrochim Acta 42:771
Frumkin AN, Melik-Gaikazyan VI (1951) Dokl Akad Nauk SSSR 77:855
Lorenz W (1958) Z Elektrochem 62:192
Armstrong RP, Rice WP, Thrisk HR (1968) J Electroanal Chem 16:517
Rammelt U, Reinhard G, Rammelt K (1980) J Electroanal Chem 180:327
Paasch G, Schwarzenberg M, Jobst K, Sawtchenko L (1990) Mater Sci Forum 62–64:455
Schneider W (1975) J Phys Chem 79:127
Ho C, Raistrick D, Huggins R (1980) J Electrochem Soc 127:343
Koppitz FD, Schultze JW, Rolle D (1984) J Electroanal Chem 170:5
Izotov VY, Strizhakova NG, Kozachkov SG, Danilin VV, Mironova AA, Maletin YA, Lust E, Arulepp M, Jänes A, Nurk G, Permann L (2003) J Power Sources (in press)
Arulepp M, Jänes A, Nurk G, Permann L, Nigu P, Lust (2000) Electrochemical properties of skeleton carbon materials with large surface area and controlled pore size. In: Abstracts of 51st Annual ISE Meeting, Warsaw, s-8, p 946
Maletin Y, Strizhakova N, Kozachkov S, Mironova A, Podmogilny S, Danilin V, Kolotilova J, Izotov YV, Cederström J, Gordeev S, Kukushkina J, Sokolov V, Kravchik A, Perkson A, Arulepp M, Leis J, Clarence LW, Zheng J (2002) Supercapacitor and a method of manufacturing such a supercapacitor. US Pat Appl 2002/0097549
Nurk G, Jänes A, Arulepp M, Nigu P, Permann L, Lust E (2001) Electric double layer structure at porous carbon electrodes. In: Meeting abstracts of joint international meeting of ISE and ECS. San Francisco, abstr 1006
Gregg SJ, Sing KSW (1982) Adsorption: surface area and porosity. Academic Press, London, p 1
Pell WG, Conway BE, Marincic N (2000) J Electroanal Chem 491:9
Lust E, Jänes A, Sammelselg V, Miidla P (2000) Electrochim Acta 46:185
Lust K, Väärtnõu M, Lust E, (2002) J Electroanal Chem 532:303
Meyers JP, Doyle M, Darling RM, Newman J (2000) J Electrochem Soc 147:2930
Nurk G, Eskusson J, Jaaniso R, Lust E (2003) J Solid State Electrochem 421:7
Taylor SR, Gileadi E (1995) Corros Sci 51:664
Sluyters-Rehbach M, Sluyters JH (1970) Sine wave methods in the study of electrode processes. In: Bard A (ed) Electroanalytical chemistry, vol 4. Dekker, New York, pp 1–127
Daikhin LI, Kornyshev AA, Urbakh M (1998) J Chem Phys 108:171
Song H-K, Jung Y-H, Lee K-H, Dao LH (1999) Electrochim Acta 44:3513
Ue M (1994) Electrochim Acta 39:2083
Gill DS (1997) Electrochim Acta 22:491
Bisquert I, Garcia-Belmonte G, Bueno P, Longo E, Bulhoes L (1998) J Electroanal Chem 452:229
Acknowledgements
This work was supported in part by the Estonian Science Foundation under project no. 4568.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lust, E., Jänes, A., Pärn, T. et al. Influence of nanoporous carbon electrode thickness on the electrochemical characteristics of a nanoporous carbon|tetraethylammonium tetrafluoroborate in acetonitrile solution interface. J Solid State Electrochem 8, 224–237 (2004). https://doi.org/10.1007/s10008-003-0396-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-003-0396-6