
Abstract
The [3+2] cycloaddition (32CA) reaction of benzonitrile oxide to α-methylene cyclopentanone and propionitrile oxide to γ-methyl-α-methylene-γ-butyrolactone, yielding regio- and stereochemically defined spiroisoxazolines, has been studied at the MPWB1K/6-311G(d,p) computational level. These processes proceed by a one-step mechanism through asynchronous transition states. Ortho regioselectivity and anti diastereofacial selectivity are predicted in complete agreement with the experimental outcomes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Spiroisoxazolines are a structurally diverse class of biologically active candidates [1,2,3,4,5,6,7,8]. Being an integral part of several natural products, such as calafianin [1, 2], aerothionin [3], agelorins [4] and psammaplysins [5], spiroisoxazolines have received special attention in organic synthesis owing to their wide therapeutic potential. The anticancer properties of the spiroisoxazoline analogues are well documented [6], while in 2017, the spiroisoxazoline SMARt-420 has been identified to revert antibiotic resistance [7], a rapidly increasing health concern. Recently, in 2019, Pratap and coworkers [8] reported the anti-proliferative and antimalarial activities of the spiroisoxazoline derivatives.
The [3+2] cycloaddition (32CA) reactions of nitrile oxides to exocyclic double bonds is the most versatile method for the synthesis of spiroisoxazolines [9]. Numerous studies [6, 9, 10] have been devoted by medicinal chemists to target spiroisoxazoline derivatives from 32CA reactions of nitrile oxides to olefins with exocyclic double bonds. These reactions yield regioisomeric spiroisoxazolines (see Scheme 1) or regio- and stereoisomeric spiroisoxazolines depending on the reagents (see Scheme 2).

Synthesis of regioisomeric spiroisoxazolines

Synthesis of regio- and stereoisomeric spiroisoxazolines
Predictability of the regio- and stereochemical outcome of these 32CA reactions depends on the careful examination of the reaction mechanism. Computational studies have come to aid to obtain a precise prediction and analysis of the mechanism, selectivity and reactivity of chemical reactions since the last two decades. Density functional theory [11] has found its place in the characterization of reactants, products, transition state structures (TSs) and intermediates on the potential energy surface (PES) [12] defined under the transition state theory [13]. Since 2000 [14], the location of TSs of several 32CA reactions have been studied in detail, and in 2017, Jasiński and coworkers performed DFT studies to locate the electronic stationary points along the PES of the 32CA reactions of benzonitrile-N-oxides and nitroethenes [15]. The MPWBIK functional has been recently stressed [16] in 2018 as a suitable system for the analysis of 32CA reactions. The reaction selectivity and activation parameters of several 32CA reactions have been examined using MPW functional [17, 18] and has provided reasonably good insight of the involved transition states. Herein, nitrile oxide cycloadditions to monosubstituted alkene derivatives with an exocyclic double bond are studied using MPW functional to understand the origin of regio- and diastereofacial selectivity in the synthesis of spiroisoxazolines. Muhlstudt et al. [19] performed nitrile oxide cycloadditions with a wide range of methylene ketones and obtained complete regioselectivity. The 32CA reaction of benzonitrile oxide, BNO 1, to α-methylene cyclopentanone, MCP 2, experimentally performed by Muhlstudt et al. [19] (see Scheme 3), has been selected as the first computational model to understand the regioselectivity.

Regioselective 32CA reaction of benzonitrile oxide, BNO 1, to α-methylene cyclopentanone, MCP 2
These 32CA reactions of propionitrile oxide with substituted methylene butyrolactones afford regioselective products with 80–100% facial selectivity in favour of the anti addition product [20]. The 32CA reaction of propionitrile oxide, PNO 4, to γ-methyl-α-methylene-γ-butyrolactone, MBL 5, experimentally performed by Paul savage et al. [20] (see Scheme 4), has been chosen as the second computational model to understand the facial selectivity in the synthesis of spiroisoxazolines.

Stereo- and regioselective 32CA reaction of propionitrile oxide, PNO 4, to γ-methyl-α-methylene-γ-butyrolactone, MBL 5
Initially, the nature of interactions between the reactants in an elementary cycloaddition step is probed from the analysis of standard global and local reactivity indices. Subsequently, all theoretically possible reaction paths for the 32CAs are examined, and the obtained energetics is analysed.
Computational methods
The Berny analytical gradient optimization method was used [21, 22] with the MPWB1K functional [23] in conjunction with the 6-311G(d,p) basis set [24]. The optimized geometries were then characterized at the same level to ensure that the reactants and products did not have any imaginary frequency and the transition states had one and only one imaginary frequency. The intrinsic reaction coordinate (IRC) [25] pathways of the investigated CAs were traced using the second order Gonzalez-Schlegel integration method [26, 27]. Solvent effects of tetrahydrofuran (THF) and benzene were taken into account using the polarizable continuum model (PCM) [28, 29] in the framework of the self-consistent reaction field (SCRF) [30,31,32]. Enthalpies, entropies and Gibbs free energies were calculated at reaction conditions 298 K (25 °C) and 1 atm in THF and benzene.
The global reactivity indices, electronic chemical potential μ and chemical hardness η, were calculated from the HOMO (EHOMO) and LUMO (ELUMO) energies using the following equations [33]:
The electrophilicity index ω is expressed in terms of the electronic chemical potential μ and chemical hardness η by the following equation [33, 34]:
The relative nucleophilicity index N [35] is expressed as
The electrophilic \( {P}_{\mathrm{k}}^{+} \) and nucleophilic \( {P}_{\mathrm{k}}^{-} \)Parr functions [36] are calculated using the following equations:
where ρsra (r) and ρsrc (r) are the Mulliken atomic spin densities of radical anion and radical cation respectively.
All computations were performed using the Gaussian 03 suite of programmes [37]. Topological analysis of the electron localization function (ELF) [38,39,40] and quantum theory of atoms in molecules (QTAIM) [41] parameters for the reactants and the TSs are also performed. These are collected in the supplementary material.
Analysis of interactions between the reactants
Conceptual density functional theory-based global and local reactivity indices [33] have been used to analyse the interactions between reactants in several biomolecular processes, especially 32CA reactions [see, e.g. 15–18]. A similar approach has also been applied in this study. The calculated global reactivity indices of BNO 1, MCP 2, PNO 4 and MBL 5 are given in Table 1. These indices are calculated at B3LYP/6-31G(d) level since the electrophilicity [34], and nucleophilicity [35] scales are defined in literature at this computational level to characterize reactants in organic reactions.
The electronic chemical potential, μ, of BNO 1, μ = − 3.81 eV, is similar to that of MCP 2, μ = − 4.00 eV, which indicates a corresponding non-polar character of the 32CA reaction, while PNO 4 shows higher electronic chemical potential, μ = − 2.91 eV, relative to MBL 5 with μ = − 4.22 eV. This predicts some polar character for the 32CA reaction of PNO 4 to MBL 5.
The electrophilicity ω indexes of BNO 1 and MCP 2 are 1.45 and 1.60 eV, respectively, BNO 1 being classified in the borderline of strong and moderate electrophiles (0.80 eV < ω < 1.50 eV) and MCP 2 being classified as strong electrophile (ω > 1.50 eV) within the electrophilicity scale [34]. PNO 4 with electrophilicity index ω = 0.57 eV is classified as a marginal electrophile while MBL 5 with ω = 1.49 eV as a strong electrophile.
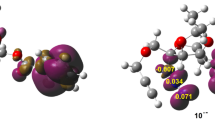
In 2004, Domingo [42] established that the asynchronicity in bond formation is controlled by the electrophilic ethylene derivative irrespective of the polar character of the 32CA reaction. Thus, the most electrophilic centre of the ethylene derivative is always involved in the formation of the first new single bond. Consequently, for 32CA reactions of BNO 1 and PNO 4 to MCP 2 and MBL 5, the electrophilic Pk+ Parr functions [36] of MCP 2 and MBL 5 were analysed (see Fig. 1). The nucleophilic Pk− Parr functions of BNO 1 and PNO 4 were also computed. The Mulliken atomic spin densities are given in Fig. 1.

Three-dimensional representation of the Mulliken atomic spin densities (isovalue = 0.0004) of radical anions 2− and 5− and radical cations 1+ and 4+ together with the electrophilic Pk+ Parr functions of MCP 2 and MBL 5 and the nucleophilic Pk− Parr functions of BNO 1 and PNO 4. Purple regions correspond to positive values, while the brown regions correspond to negative regions of the Mulliken atomic spin densities
For the C4–C5 bond, the electrophilic Pk+ Parr functions of C4 and C5 centres in MCP 2, 0.50 and 0.01, respectively, and in MBL 5, 0.58 and 0.09, respectively, indicate that the first single bond will involve the C4 carbon. Finally, the O1 oxygen of BNO 1 and PNO 4 with Pk− = 0.45 and 0.67, respectively, presents the most nucleophilic activation, while the C3 is lesser nucleophilically activated in BNO 1 with Pk− = 0.02 and in PNO 4 with Pk− = 0.28.
Analysis of the energy profile and geometry of transition states
32CA reaction of benzonitrile oxide BNO 1 to α-methylene cyclopentanone MCP 2
Due to the non-symmetry of methylene cyclopentanone MCP 2, two regioisomeric reaction paths, labelled ortho and meta (see Scheme 5), are feasible for this 32CA reaction. The ortho reaction path is associated with the formation of O1–C5 and C3–C4 bonds, while the meta channel involves formation of O1–C4 and C3–C5 bonds. Stationary points along these four reaction paths were searched in this study, which allowed locating and characterizing the reagents, BNO 1 and MCP 2, two transition states, i.e. TS1 and TS2, and the corresponding cycloadducts CA1 and CA2 (see Scheme 5). Cycloadduct CA1 is the ortho product, and CA2 is the meta product. This 32CA reaction follows one-step mechanism. The relative energies, enthalpies and free energies of products and TSs in gas phase as well as THF are given in Table 2.

Studied reaction paths of the 32CA reaction of BNO 1 to MCP 2
Analysis of the relative energies leads to some appealing conclusions. The activation energies are 11.4 kcal mol−1 (TS1) and 15.9 kcal mol−1 (TS2) in gas phase and 13.4 kcal mol−1 (TS1) and 18.1 kcal mol−1 (TS2) in THF, with the 32CA reaction being strongly exothermic with 48.3 kcal mol−1 (CA1) and 43.4 kcal mol−1 (CA2) in gas phase and 46.4 kcal mol−1 (CA1) and 41.6 kcal mol−1 (CA2) in THF (see Table 2). The inclusion of THF increases the activation enthalpies by 2.3 kcal mol−1 (TS1) and 2.6 kcal mol−1(TS2) as a consequence of a larger solvation of the reagents than TSs [43]. The activation energy 11.4 kcal mol−1 associated with TS1 is lower than that associated with the non-polar 32CA reaction of BNO 1 with methyl acrylate, 12.3 kcal mol−1, in which the ortho TS is lower than meta TS by only 1.6 kcal mol−1 [44]. For the 32CA reaction of BNO 1 and MCP 2, the most favourable reaction path is associated with the ortho approach mode, yielding the cycloadduct CA1 via TS1 in complete agreement with the experimental results [19]. This 32CA reaction is completely regioselective, as TS1 is 4.5 kcal mol−1 lower in activation energy than TS2 in gas phase and by 4.7 kcal mol−1 in THF. The formation of product CA1 is strongly exothermic which makes the reaction irreversible.
Thermal corrections to the electronic energies give the relative enthalpies in gas phase and THF. The activation enthalpies increase by 0.4 and 0.6 kcal mol−1 in gas phase and 0.7 and 1.0 kcal mol−1 in THF relative to the activation energies, while reaction enthalpies decrease by 2.6 and 3.0 kcal mol−1 in gas phase and 3.1 and 3.4 kcal mol−1 in THF relative to the reaction energies. Inclusion of the entropies to enthalpies strongly increases the activation Gibbs energies by 13.2 and 14.3 kcal mol−1 in gas phase and THF, while the reaction Gibbs energies show a sharp decrease by 14.3 and 15.1 kcal mol−1 in gas phase and 14.1 and 15.0 kcal mol−1 in THF.
The activation Gibbs free energy of cycloadduct CA1 becomes 25.0 kcal mol−1 in gas phase and 27.3 kcal mol−1 in THF, the 32CA reaction being strongly exergonic by 31.4 kcal mol−1 in gas phase and 29.2 kcal mol−1 in THF. Figure 2 shows the optimized geometries of the TSs.

MPWB1K/6-311G(d,p) optimized transition states for 32CA reaction of BNO 1 and MCP 2. Bond lengths are given in angstrom units. Values in parenthesis indicate the bond lengths calculated in THF
Now, formation of C–C and C–O single covalent bonds begin at a distance of 2.0–1.9 Å and 1.7–1.6 Å [17] respectively, which indicates that the formation of new C–C and C–O covalent bonds has not started in the TSs with C–C and C–O forming bond distances greater than 2.0 Å (see Fig. 2). The forming C–C bond length is shorter than C–O forming bond length at TS1, while the shorter bond length corresponds to the formation of C–O forming bond at TS2. These two TSs show almost similar asynchronicity, and inclusion of the solvent effects of THF slightly changes the forming bond lengths at these two TSs.
32CA reaction of propionitrile oxide PNO 4 to γ-methyl-α-methylene-γ-butyrolactone MBL 5
Due to the non-symmetry of both the reagents PNO 4 and MBL 5, different stereo- and regioisomeric paths exist for this 32CA reaction. The two regioisomeric reaction paths labelled ortho and meta and the two diastereofacial isomeric reaction paths, labelled syn and anti, associated with the participation of nitrile oxide and exocyclic double bond addition have been considered here (see Scheme 6). The ortho regioisomeric channel is associated with the formation of C3–C4 and C5–O1 bonds, while the meta path involves formation of C3–C5 and C4–O1 bonds. The anti diastereofacial reaction path is associated with the addition opposite to the methyl substituent, while the syn diastereofacial reaction path is associated with the addition to the same face of the methyl substituent as coined by Savage and coworkers [20] for this 32CA reaction.

Studied reaction paths of the 32CA reaction of propionitrile oxide PNO 4 to γ-methyl-α-methylene-γ-butyrolactone MBL 5
Along these four reaction paths, four TSs, i.e. TS3, TS4, TS5 and TS6, were located and characterized and the corresponding cycloadducts, i.e. CA3, CA4, CA5 and CA6. This 32CA reaction follows a one-step mechanism, and the relative energies in gas phase and benzene are given in Table 2. The activation energies range from 11.4 (TS3) to 15.8 (TS6) kcal mol−1 in gas phase and from 12.6 (TS3) to 17.7 (TS5) kcal mol−1 in benzene, with this 32CA reaction being strongly exothermic from − 44.2 (CA5) to − 48.3 (CA3) kcal mol−1 in gas phase and − 42.5 (CA5) to − 46.6 (CA3) kcal mol−1 in benzene. Inclusion of solvent effects in benzene increases the activation enthalpy by 1.3–2.2 kcal mol−1 as a consequence of a larger solvation of the reagents than TSs [44]. This 32CA reaction is completely ortho regioselective, as TS5 and TS6 are higher in energy than TS3 by 4.2 and 4.4 kcal mol−1 in gas phase and 5.1 and 5.0 kcal mol−1 in benzene. The activation energy of TS4 is higher in energy than TS3 by 1.1 kcal mol−1 in gas phase and by 1.2 kcal mol−1 in benzene, which accounts for the 81:19 diastereomeric ratio of ortho/anti and ortho/syn products obtained experimentally in benzene at room temperature [20].
The inclusion of thermal corrections to the electronic energies causes minimal increase in activation enthalpies by 0.3–0.5 kcal mol−1, while the reaction enthalpies are increased by 2.5–3.1 kcal mol−1. The activation Gibbs free energies are increased by between 12.9 and 14.1 kcal mol−1 relative to the activation enthalpies, while the reaction Gibbs energies are strongly increased by between 14.4 and 15.0 kcal mol−1, which is due to the unfavourable entropies associated with this 32CA process. The relative Gibbs free energies clearly account for the complete ortho regioselectivity and anti diastereofacial selectivity experimentally observed [20].
The optimized geometries of four TSs are given in Fig. 3.

MPWB1K/6-311G(d,p) optimized transition states for 32CA reaction of PNO 4 and MBL 5. Bond lengths are given in angstrom units. Values in parenthesis indicate the bond lengths calculated in benzene
At the ortho TSs, TS3 and TS4, the length of forming C5–O1 bond is greater than that of the forming C3–C4 bond, while at the meta TSs, TS5 and TS6, the length of forming C3–C5 bond is greater than that of the forming C4–O1 bond. Forming C3–C4 and C5–O1 bond distances greater than 2.0 Å indicates that no covalent bond formation has begun yet in any of the four TSs. Meta and ortho TSs show almost similar asynchronicity. Inclusion of solvent effects in benzene scarcely changes the forming bond lengths.
Conclusions
The 32CA reactions of α-methylene cyclopentanone and γ-methyl-α-methylene-γ-butyrolactone to benzonitrile oxide and propionitrile oxide, yielding spiroisoxazolines with regio- and stereoselective control, have been studied at MPWB1K/6-311G(d,p) level of theory. Analysis of Parr functions indicates the initiation from the unsubstituted carbon atom of the ethylene derivative. These 32CA reactions present activation enthalpy of 11.8 and 11.7 kcal mol−1 in gas phase, while 14.1 and 12.9 kcal mol−1 are the respective calculated values in THF and benzene. Analysis of the activation Gibbs free energies indicates that these 32CA reactions are completely ortho regioselective and the second reaction proceeds through anti diastereofacial selectivity under kinetic control, in complete agreement with the experimental findings.
References
Encarnacion RD, Sandoval E, Malmstrom J, Christophersen C (2000) Calafianin, a bromotyrosine derivative from the marine sponge Aplysina gerardogreeni. J Nat Prod 63:874–875. https://doi.org/10.1021/np990489d
Bardhan S, Schmitt DC, Porco JA (2006) Total synthesis and stereochemical assignment of spiroisoxazoline natural product (+) calafianin. Org Lett 8:927–930. https://doi.org/10.1021/ol053115m
Ogamino T, Obata R, Nishiyama S (2006) Asymmetric synthesis of aerothionin, a marine dimeric spiroisoxazoline natural product, employing optically active spiroisoxazoline derivative. Tetrahedron Lett 47:727–730. https://doi.org/10.1016/j.tetlet.2005.11.097
Konig GM, Wright AD (1993) Agelorins A and B, and 11-epi-fistularin-3,three new antibacterial fistularin-3 derivatives from the tropical marine sponge Agelas oroides. Heterocycles 36:1351–1358. https://doi.org/10.3987/COM-92-6317
Perron F, Albizati KF (1989) Chemistry of spiroketals. Chem Rev 89:1617–1661. https://doi.org/10.1021/cr00097a015
Khazir J, Singh PP, Reddy M, Hyder I, Shafi S, Sawant SD, Chashoo G, Mahajan A, Alam MS, Saxena AK, Arvinda S, Gupta BD, Sampath Kumar HM (2013) Synthesis and anticancer activity of novel spiro-isoxazoline and spiro-isoxazolidine derivatives of α-santonin. Eur J Med Chem 63:279–289. https://doi.org/10.1016/j.ejmech.2013.01.003
Blondiaux N, Moune M, Desroses M, Frita R, Flipo M, Mathys V, Soetaert K, Kiass M, Delorme V, Djaout K, Trebosc V, Kemmer C, Wintjens R, Wohlkönig A, Antoine R, Huot L, Hot D, Coscolla M, Feldmann J, Gagneux S, Locht C, Brodin P, Gitzinger M, Déprez B, Willand N, Baulard AR (2017) Reversion of antibiotic resistance in mycobacterium tuberculosis by spiroisoxazoline SMARt-420. Science 335:1206–1211. https://doi.org/10.1126/science.aag1006
Pratap S, Naaz F, Reddy S, Jha KK, Sharma K, Sahal D, Akhter M, Nayakanti D, Kumar HMS, Vandana PK, Shafi S (2019) Anti-proliferative and anti-malarial activities of spiroisoxazoline analogues of artemisinin. Arch Pharm 352:1800192. https://doi.org/10.1002/ardp.201800192
Savage GP (2010) Spiro isoxazolines via nitrile oxide 1,3-dipolar cycloaddition reactions. Curr Org Chem 14:1478–1499. https://doi.org/10.2174/138527210791616812
Zaki M, Oukhrib A, Akssira M, Berteina-Raboin S (2017) Synthesis of novel spiro-isoxazoline and spiro-isoxazolidine derivatives of tomentosin. RSC Adv 7:6523–6529. https://doi.org/10.1039/C6RA25869G
Parr RG, Yang W (1989) Density-functional theory of atoms and molecules, vol 4. Oxford University Press, Oxford, pp 70–86
Moss SJ, Coady CJ (1983) Potential-energy surfaces and transition state theory. J Chem Educ 60:455–461. https://doi.org/10.1021/ed060p455
Trautz M (1916) Das gesetz der reaktionsgeschwindigkeit und der gleichgewichte in gasen. Bestätigung der additivität von Cv-3/2R. Neue bestimmung der integrationskonstanten und der moleküldurchmesser. Z Anorg Allg Chem 96:1–28
Carda M, Portolés R, Murga J, Uriel S, Marco JA, Domingo LR, Zaragozá RJ, Röper H (2010) Stereoselective 1,3-dipolar cycloadditions of a chiral nitrone derived from erythrulose. An experimental and DFT theoretical study. J Org Chem 65:7000–7009. https://doi.org/10.1021/jo0009651
Jasiński R, Jasińska E, Dresler E (2017) A DFT computational study of the molecular mechanism of [3 + 2] cycloaddition reactions between nitroethene and benzonitrile N-oxides. J Mol Model 23:13. https://doi.org/10.1007/s00894-016-3185-8
Domingo LR, Ríos-Gutiérrez M, Pérez P (2018) A molecular electron density theory study of the reactivity and selectivities in [3+2] cycloaddition reactions of C,N-dialkyl nitrones with ethylene derivatives. J Org Chem 83:2182–2197. https://doi.org/10.1021/acs.joc.7b03093
Ríos-Gutiérrez M, Domingo LR (2019) Unravelling the mysteries of the [3+2] cycloaddition reactions. Eur J Org Chem:267–282. https://doi.org/10.1002/ejoc.201800916
Domingo LR, Acharjee N (2018) [3+2] Cycloaddition reaction of C-phenyl-N-methyl nitrone to acyclic-olefin-bearing electron-donating substituent: a molecular electron density theory study. ChemistrySelect 3:8373–8380. https://doi.org/10.1002/slct.201801528
Muller G, Frischleder H, Muhlstadt M (1969) Mono- und bicyclische _- methylenketone als dipolarophile. J Prakt Chem 311:118–129. https://doi.org/10.1002/prac.19693110117
Pereira SM, Savage GP, Simpson GW, Greenwood RJ, Mackay MF (1993) Diastereoselective propionitrile oxide cycloaddition reactions with some γ-substituted α-methylene-γ-butyrolactones. Aust J Chem 46:1401–1412. https://doi.org/10.1071/CH9931401
Schlegel HB (1982) Optimization of equilibrium geometries and transition structures. J Comput Chem 3:214–218. https://doi.org/10.1002/jcc.540030212
Schlegel HB (1994) Modern electronic structure theory. World Scientific Publishing, Singapore
Zhao Y, Truhlar DG (2004) Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: the MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J Phys Chem A 108:6908–6918. https://doi.org/10.1021/jp048147q
Hehre WJ, Radom L, Schleyer PVR, Pople JA (1996) Ab initio molecular orbital theory. Wiley, New York. https://doi.org/10.1002/jcc.540070314
Fukui K (1970) Formulation of the reaction coordinate. J Phys Chem 74:4161–4163. https://doi.org/10.1021/j100717a029
González C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527. https://doi.org/10.1021/j100377a021
González C, Schlegel HB (1991) Improved algorithms for reaction path following: higher-order implicit algorithms. J Chem Phys 95:5853–5860. https://doi.org/10.1063/1.461606
Tomasi J, Persico M (1994) Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem Rev 94:2027–2094. https://doi.org/10.1021/cr00031a013
Simkin BY, Sheikhet II (1995) Quantum chemical and statistical theory of solutions: a computational approach. Ellis Horwood, London
Cossi M, Barone V, Cammi R, Tomasi J (1996) Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem Phys Lett 255:327–335. https://doi.org/10.1016/0009-2614(96)00349-1
Cances E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107:3032–3041. https://doi.org/10.1063/1.474659
Barone V, Cossi M, Tomasi J (1998) Geometry optimization of molecular structures in solution by the polarizable continuum model. J Comput Chem 19:404–417. https://doi.org/10.1002/(SICI)1096-987X(199803)19:4<404::AID-JCC3>3.0.CO;2-W
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21:748 (22 pages). https://doi.org/10.3390/molecules21060748
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 58:4417–4423. https://doi.org/10.1016/S0040-4020(02)00410-6
Domingo LR, Pérez P (2011) The nucleophilicity N index in organic chemistry. Org. Biomol Chem 9:7168–7175. https://doi.org/10.1039/C1OB05856H
Domingo LR, Pérez P, Sáez JA (2013) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3:1486–1494. https://doi.org/10.1039/C2RA22886F
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision D.01. Gaussian, Inc., Wallingford CT
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92:5397–5403. https://doi.org/10.1063/1.458517
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371:686–686. https://doi.org/10.1038/371683a0
Yepes D, Murray JS, Pérez P, Domingo LR, Politzer P, Jaque P (2014) Complementarity of reaction force and electron localization function analyses of asynchronicity in bond formation in Diels–Alder reactions. Phys Chem Chem Phys 16:6726–6734. https://doi.org/10.1039/C3CP54766C
Bader RFW (1990) Atoms in molecules: a quantum theory. Clarendon Press, Oxford
Aurell MJ, Domingo LR, Pérez P, Contreras R (2004) A theoretical study on the regioselectivity of 1,3-dipolar cycloadditions using DFT-based reactivity indexes. Tetrahedron 60:11503–11509. https://doi.org/10.1016/j.tet.2004.09.057
Benchouk W, Mekelleche SM, Silvi B, Aurell MJ, Domingo LR (2011) Understanding the kinetic solvent effects on the 1,3-dipolar cycloaddition of benzonitrile N-oxide: a DFT study. J Phys Org Chem 24:611–618. https://doi.org/10.1002/poc.1858
Ndassa IM, Adjieufack AI, Ketcha JM, Berski S, Ríos-Gutiérrez M, Domingo LR (2017) Understanding the reactivity and regioselectivity of [3+2] cycloaddition reactions between substituted nitrile oxides and methyl acrylate. A molecular electron density theory study. Int J Quantum Chem 117:e25451. https://doi.org/10.1002/qua.25451
Acknowledgments
The author is thankful to Professor Manas Banerjee, Retired Professor, at The University of Burdwan, India, for his kind cooperation. The author also acknowledges the help and support of Professor Luis R Domingo, Professor, at the University of Valencia, Spain, for important clarifications in several studies related to the concept of 32CA reactions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
Tables with the MPWB1K/6-311G(d,p) electronic energies, enthalpies and Gibbs free energies of the stationary points involved in the 32CA reactions of nitrile oxides 1 and 4 with MCP 2 and MBL 5, in gas phase, THF and benzene. ELF topological analysis at the reactants and TSs and calculations of QTAIM parameters. (DOCX 1124 kb).
Rights and permissions
About this article

Cite this article
Acharjee, N. Theoretical analysis of the regio- and stereoselective synthesis of spiroisoxazolines. J Mol Model 26, 117 (2020). https://doi.org/10.1007/s00894-020-04372-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-04372-x