
Abstract
Escalating demand for sustainable energy resources, because of the rapid exhaustion of conventional energy resources as well as to maintain the environmental level of carbon dioxide (CO2) to avoid its adverse effect on the climate, has led to the exploitation of photovoltaic technology manifold more than ever. In this regard organic materials have attracted great attention on account of demonstrating their potential to harvest solar energy at an affordable rate for photovoltaic technology. 2-vinyl-4,5-dicyanoimidazole (vinazene) is considered as a suitable material over the fullerenes for photovoltaic applications because of its particular chemical and physical nature. In the present study, DFT approaches are employed to provide an exposition of optoelectronic properties of vinazene molecule and molecular crystal. To gain insight into its properties, different forms of exchange correlation energy functional/potential such as LDA, GGA, BLYP, and BL3YP are used. Calculated electronic structure of vinazene molecule has been displayed via HOMO-LUMO isosurfaces, whereas electronic structure of the vinazene molecular crystal, via electronic band structure, is presented. The calculated electronic and optical properties were analyzed and compared as well. Our results endorse vinazene as a suitable material for organic photovoltaic applications.

Vinazene molecule and molecular crystalᅟ
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Repaid demand for sustainable energy resources has increased more than ever because of threatening catastrophic changes in global climate by emission of greenhouse gases from conventional energy resources via combustion process. In addition, conventional resources of energy have speedily fallen. Therefore extensive efforts are being done to search for such alternate energy systems which can fulfill future energy requirements alongside stabilizing the atmospheric CO2 level to preserve a friendly environment [1, 2]. In this regard photovoltaic systems are considered to be perfect candidates. Photovoltaic is the technology that harvests the solar energy by converting the solar radiations directly into electricity. Because of having huge potential of supplying approximately 104 times larger energy than our present needs, solar energy is expected to be the best solution for the problem [3, 4].
Global industrial sectors have progressed significantly in exploitation of photovoltaic technologies that relied largely upon inorganic materials like silicon over a decade ago. Though inorganic materials have shown their performance well in photovoltaic devices, several disadvantages in terms of high cost and limited resources have sparked the interest among researchers to explore and investigate the replacement of inorganic materials. Beside inorganic materials, from the beginning of the twenty first century, organic materials have also shown ample potential for photovoltaic (PV) systems [5–7]. Therefore, in the present era, considerable attention is being focused on organic materials in order to search for potential systems as base materials in photovoltaic systems and solar cell devices. Moreover, because of offering some crucial advantages such as: low cost, light, flexible, thin, availability in abundance, sustainability, utilization in large area and easy manufacturing, organic photovoltaic (OPV) systems have received extraordinary scientific and industrial attention very recently [8–10]. In this regard fullerenes solar cells have demonstrated reasonable efficiency up to 11 %. However, the unique chemical and physical nature of 2-vinyl-4,5-dicyanoimidazoles (vinazene) has demonstrated a great potential for OPV applications, and has fascinated both the academic sector and industrial communities. Moreover, it has received considerable attention due to its high stability, high solubility, and high efficiency besides its suitability as an electron acceptor material [11, 12]. However, to the best of our knowledge only one theoretical study has ever been reported on vinazene so far [13], compared to the excessive experimental works that was previously reported [14, 15]. In the present work, we make a step ahead to report an extensive and comparative study, by employing first principles approaches based on the density functional theory (DFT), related to the molecule and molecular crystal of vinazene.
The right account of the electronic band structure of a specific material plays a crucial role to endorse it for a particular application. In this paper, we present an intensive theoretical study of the electronic band structure and corresponding optoelectronic properties for vinazene in both forms: molecule and molecular crystal. This work is performed by applying state of the art first principles quantum mechanical approach implemented within DMol3 [16, 17] and vienna ab-initio simulation package (VASP) [18]. As a first step, structures of the molecule and molecular crystal of vinazene are optimized by relaxing the atomic positions at the level of different exchange correlation energy functionals like LDA, GGA, BLYP, and B3LYP and then, comprehensively, their optoelectronic properties are computed. Throughout the discussion, we focus on the bridge between the optoelectronic structure and the possible OPV application.
Computational details and description of structures
Vinazene molecule structure was investigated using DFT based method, implemented in DMol3 computational code [16, 17]. This was carried out using different local and non local exchange correlation energy functional/potential such as LDA [19], GGA [20], BLYP [21], and B3LYP [22]. In this scheme of calculations, molecular orbitals are expanded in terms of linear combination of atomic orbitals/atomic basis functions and the atomic orbital basis set. The atomic orbital basis set is derived numerically on an atomic centered grid using “double numeric plus polarization” (DNP) numerical functions. To attain an adequate accuracy, a radial cutoff of 3.7 Å is used for all calculations. To depict core states, “all electrons” were taken into account [23]. Total energy convergence criteria for self-consistent field (SCF) were set to 10−6 eV. To improve computational performance in terms of fast SCF convergence, a smearing of 0.005 Hartree was considered. Computations were carried out at the gamma point of the first Brillouin zone. Regarding the molecular crystal of vinazene, we used the VASP code [18–20]. Plane wave cutoff energy of 520 eV was used for the cell optimization. Electronic iterations convergence was considered 10−5 eV using the normal (blocked Davidson) algorithm and reciprocal space projection operators. K-spacing was set 0.5 per Å, leading to a 2x1x2 mesh. The k-mesh is forced to be centered on Γ-point. To simulate the crystal structure of vinazene, experimentally obtained crystal data, for lattice parameters, were used as a starting point. Then, a full geometry of the modeled unit cell of vinazene molecular crystal structure, containing 120 atoms, was optimized using conjugate gradient algorithm. Consequently, all the atomic coordinates as well as cell parameters of the simulated molecular crystal structure were relaxed without applying any symmetry constraint. Correspondingly the energy between successive iterations was found to be less than 10−3 eV and force value 0.03 eV/Å.
Results and discussion
Optimized vinazene molecule structure and corresponding molecular crystal are shown in Fig. 1. Generally, the molecule of vinazene is made up from an aromatic imidazole ring. The aromatic imidazole ring exists in two tautomeric forms with the hydrogen atom moving between two nitrogen atoms. Each rings consists of nitrogen atoms at positions 1 and 3, hydrogen atom at position 1, a vinyl group at position 2, carbon atoms at positions 2, 4, and 5, and a pair of cyano substituent at positions 4 and 5 [24]. In this work, we presented the nitrogen atoms, carbon atoms, and hydrogen atoms in different colors: blue, gray and white respectively. We also show the skeletal diagram of the molecule that has proposed by Rasmussen et al. [25] who sparked the main interest among researchers to study this material by predicting its potential implication in modern technologies especially in photovoltaic systems.

Molecule (a), molecular crystal (b) and skeletal diagram (c) of vinazene
Electronic structure, HOMO-LUMO isosurfaces and band gap
Electronic properties play an important role in understanding the performance of OPV materials. As a matter of fact, electronic properties are mostly determined by molecular energy level near the Fermi energy. Gap between the energy level around the Fermi level is defined as energy band gap. Here, the molecular energy gaps for the molecule refers to the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Whereas energy gap for the molecular crystal refers to the energy gap between minimum of the conduction and maximum of the valence band. Smaller energy band gap OPV materials can show better efficiency as a result of higher electron excitation probability. Thus, to emphasize the possible OPV application, the electronic structure of the single molecule and molecular crystal of vinazene are investigated and discussed.
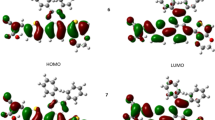
We started with the vinazene molecule, where all calculations of electronic properties were performed using DMol3 package. Different exchange correlation energy functional approximations such as LDA, GGA, BLYP, and B3LYP are used in order to see their impact on the energy band gap values. In order to obtain the energy band gap values, HOMO-LUMO of the material is desired, since HOMO-LUMO defines reactivity of the molecular material [26]. Therefore, in the first part, calculations were performed to obtain HOMO and LUMO isosurfaces of the vinazene molecule and the obtained respective isosurfaces (derived from the band structures at X and Γ points) are shown in Fig. 2.

HOMO-LUMO of vinazene molecular crystal (a) and isolated molecule (b)
From the HOMO-LUMO isosurfaces of the vinazene molecule, the energy gap values, evaluated corresponding to each exchange and correlation functional has been gathered in Table 1. We may observe that calculations using LDA exchange-correlation [19] potential [19] on vinazene molecule with parameterizations of VWN and PWC give the energy gap values of 3.282 and 3.283 eV respectively in comparison with other approximations. These energy band gap values could be interesting for technological applications, however it is better to recalculate the energy gap by applying other exchange-correlation energies since LDA approach is known for showing its tendency for over binding. As a result (either molecular crystal or molecule) calculated band gap values at level of LDA are usually underestimated. Thus, calculations of energy gap values at level of different exchange-correlation functionals are very important for predicting reliable energy gap of any material. Moreover, successful application of GGA functionals [20, 21, 27–29] in improving accuracy in the calculation of electronic structure has been reported for different materials in literature, especially with hybrid functional [30]. By taking into account advantages of GGA functional, and in order to make comparisons between LDA and other exchange correlation approaches, we adopted GGA approach with seven different parameterizations as listed in Table 1. Results obtained for energy gap values within different approximations are in the range 3.284 to 3.336 eV. One can see that all these values are consistent/nearly the same although various parameterizations were used. This indicates that the energy gap calculations with GGA approximations are almost independent from chosen parameterizations. From Table 1, one can see the slight difference between results obtained with GGA in comparison to LDA band gap value.
In fact, hybrid functionals are widely used in the molecular studies due to their improved incorporation of the missing features from LDA and GGA functionals [30]. The improvement of the functional has lead to better results in total energies and energy gap of electronic structure. In addition, these functionals are also highly recommended to study the phenomena of charge transfer, especially where the exchange interaction is an important aspect to evaluate [31]. B3LYP [22] is one of the most updated hybrid Hartree-Fock (HF) density functionals, that combines Becke’s three parameter exchange functional [32] with Lee-Yang-Parr’s correlation functional [33]. This updated functional version of BLYP has been reported to be successful in providing good results for structural and excited states properties [31]. In order to complement our energy gap calculations of vinazene molecule with LDA and GGA, we also carried out calculations by applying B3LYP and BLYP hybrid functionals. Calculations of the electronic structure with BLYP hybrid functional reflect energy gap value of 3.274 eV. However with more updated hybrid functional of B3LYP, obtained energy gap value has been drastically increased, i.e., 4.675 eV.
Similar to the vinazene molecule, electronic properties calculations on vinazene molecular crystal have also been performed. Figure 3 shows the obtained energy spectra near the Fermi level of vinazene molecular crystal that was calculated by applying exchange correlation energy of GGA [20]. GGA functional is chosen due to its abilities in computing good results for organic molecules especially for vinazene structure. As shown in Fig. 3, the lowest conduction band lies at the X point with an energy value of 2.9388 eV and the highest valence band lies at the Γ point with an approximate energy value of 0 eV with reference to the Fermi level. These values clearly indicate the high oxidative stability of vinazene molecular crystal, which is a key requirement for an organic electronic material. It also shows, that vinazene molecular crystal exhibits an indirect band gap. So, a change of electron momentum occurs to allow the transition of electrons from valence to conduction band. Since there is a change in momentum, the electron has to occupy more energy to be diffused in the conduction region. This result clearly shows the energy required must be larger than the energy gap value of Eg = 2.9388 eV. Fortunately, the obtained energy gap is considered small for a basic structure of crystalline OPV material compared to the others OPVs. Consequently it may be deduced that vinazene molecular crystal possess the suitable characteristic as a good electron acceptor material and contains a high ability in conducting electrons with holes.

Zoomed energy spectra of vinazene molecular crystal
Optical properties
In addition to the electronic properties, we have also calculated the optical properties using VASP package [18]. All of the optical parameter calculations are based on DFT and were done by applying the exchange correlation functional of GGA with adopted PBE parameterization. The calculated optical properties of reflectivity, refractive index, n, absorption coefficient, k, together with real and imaginary parts of the frequency dependent dielectric and conductivity function, for vinazene are shown in Figs. 4, 5, and 6. These calculations were done for both molecule and molecular crystal forms.

Reflectivity spectra of vinazene: isolated molecule (a) and molecular crystal (b). They are presented in black, red, and blue color lines, respective to three polarization vectors of (100), (010), and (001) directions

Refractive index n and extinction coefficient k versus energy. They are presented in black, red, and blue color lines, respective to three polarization vectors of (100), (010), and (001) directions

Real and imaginary parts (ε 1 and ε 2) of dielectric constants and real and imaginary parts (σ1 and σ2) of conductivity function for vinazene molecular crystal at the right side and molecule at the left side. They are presented in black, red, and blue color lines, respective to three polarization vectors of (100), (010), and (001) directions
Figure 4 shows the reflectivity of the molecule and molecular crystal of vinazene. It can be seen from these graphs, the highest peaks were contributed from the (010) direction and interestingly, reflectivity of both graphs start to reach a plateau after 30 eV. In the energy range of 0 to 5 eV of vinazene molecular crystal, maximum reflectivity from (001), (010), and (001) directions occurs with a value of 0.506. Whereas the maximum reflectivity value of vinazene molecule is only 0.08 which occurs in the energy range of [5–10] eV and [14–16] eV. Obtained value of reflectivity for vinazene molecule is greatly lower compared to vinazene molecular crystal. When the reflectivity value is low, it generally shows that level of transparency of the material is high. Thus, vinazene molecule is more efficient to play the role of OPV material compared to its molecular crystal since it is almost transparent for the photon energy less than 50 eV.
Besides reflectivity spectra, it is important to highlight the function of refractive index and extinction coefficient in the calculation of optical properties. By referring to Fig. 4, the refractive index for both vinazene molecule and molecular crystal showed a similar pattern with different level of indices. The highest refractive index of vinazene molecule at 0 eV is 1.2 which is lower than vinazene crystal by 0.8. This means that vinazene crystal have higher tendency to refract photon compared to vinazene molecule. Both of these structures have high indices values in energy range of [0–10] eV; therefore the generated outputs in the respective region are quite low. However, the situation of low generated outputs is turned over, starting from the energy range of 10 eV. The refractive index graphs clearly show both of the materials are able to generate high outputs with the input photon energy as low as 10 eV. Interestingly, from the energy of 20 eV onward both structures continuously show stable indices with a value of 1.0.
As the refractive index function reported above, the absorption coefficient function is also discussed in parallel. A similar pattern for both vinazene molecular crystal and molecule is also seen in the absorption coefficient spectra graphs. In the energy range of [0–10] eV, the highest absorption peak values for vinazene molecule and molecular crystal are 0.6 and 2.6 respectively. The calculated absorption value for vinazene molecular crystal is higher compared to vinazene isolated molecule due to the arrangement and size of the structure system. A larger structure will have larger windows to harvest light and consequently contributes to higher intensity of spectrum absorbance. The absorption spectra of both structures cover ultraviolet spectrum and extends to the infrared zone. This shows the vinazene can absorb a wide range of electromagnetic spectrum with suitable arrangement. The performance of vinazene crystal was also found to be more efficient than vinazene molecule since vinazene crystal has achieved its stability in the transition process starting from the energy of 10 eV. However, the vinazene molecule starts to stabilize only on the energy of 20 eV.
Furthermore, Fig. 5 shows the real and imaginary part of the frequency dependent dielectric and conductivity function for vinazene molecular crystal and molecule. Significant differences are found in the dielectric spectra for both forms. All of the polarization vectors for vinazene molecule exhibit an intense spectrum in the energy below 20 eV. Whereas for vinazene crystal, only polarization vectors of (100) and (010) directions tend to have a slight intense spectra, but it only occurs in the energy below 10 eV. The obtained values for the real part of dielectric function for vinazene molecule at 0 eV are ε(100) = 1.4486, ε(010) = 1.4206, and ε(001) = 1.2903 and for vinazene crystal are ε(100) = 4.1788, ε(010) = 4.1989, and ε(001) = 2.6910. These values might suggest that vinazene crystal has a great potential to play a role as an electron acceptor material as compared to vinazene molecule. This is due to the higher dielectric constant values which indirectly results in a high capability in conducting and transporting the electrons.
Almost similar optical spectra are also found in Fig. 5 for the real and imaginary parts of conductivity function. The real part of conductivity function for vinazene molecule is clearly monopolized by the (001) polarization vectors, while the imaginary parts are evenly distributed for all polarization vectors. This is different from real and imaginary parts of the molecular crystal, where the majority of the spectra values are contributed from the (010) directions. In the energy range of [0–20] eV, the real part of conduction spectrum for both form shows a drastic increase but a sudden decrease appears after it reaches a plateau. The plateau represents the achievement of optimum stability of the structure that indirectly results to a high capability in conducting and transporting electrons smoothly.
Conclusions
We have successfully emphasized the understanding of optoelectronic properties of vinazene molecule and molecular crystal as an effective molecular material for organic photovoltaic applications. Systematic studies of electronic and optical properties were accomplished using density functional theory computational approaches. Several exchange correlation functionals were employed to attain a sturdy calculations result. Our study has further opened the possibility of performing systematic computational investigations of the corresponding electronic and optical response. In addition, by establishing HOMO and LUMO of vinazene molecule and band structure of the corresponding molecular crystal, reliable energy band gap values have been evaluated. From a deep analysis of energy band gaps, reflectivity, refractive index, absorption coefficient, dielectric, and conductivity function of vinazene molecule and molecular crystal, we were able to accurately predict the possibility of integrating vinazene in OPV devices and discussing its performance. Hence, we came to a conclusion that vinazene molecular crystal is more compatible, compared to the molecule form, to serve as a new active material for applications in the OPV system. Furthermore, DFT approach is found to be a good predictive tool for the study of excitation energy of molecular material. Because of unavailability of experimental measurements and not very many reported studies related to this material, our study egg on researchers for further investigations on this potential OPV material for energy harvesting to realize the goal of green and sustainable energy technologies.
References
Goffman E (2008) ProQuest
Dahl S, Chorkendorff I (2012) Nat Mater 11:100
Polman A, Atwater HA (2012) Nat Mater 11:174
Li G, Shrotriya V, Huang J, Yao Y, Moriarty T, Emery K, Yang Y (2005) Nat Mater 4:864
Fan Z, Javey A (2008) Nat Mater 7:835–836
Liu Z, Xu J, Chen D, Shen G (2015) Chem Soc Rev 44:161
Guo X, Baumgarten M, Müllen K (2013) Prog Polym Sci 38:1832
Ryyan Khan M, Ray B, Alam MA (2014) Solar Energy Mater Solar Cells 120 Part B(716)
Zhao G, He Y, Li Y (2010) Adv Mater 22:4355
Stratakis E, Stylianakis MM, Koudoumas E, Kymakis E (2013) Nanoscale 5:4144
Woo CH, Holcombe TW, Unruh DA, Sellinger A, Frechet JMJ (2010) Chem Mater 22:1673
Lim E, Lee S, Lee KK (2012) Mol Cryst Liq Cryst 565
Bloking JT, Han X, Higgs AT, Kastrop JP, Pandey L, Norton JE, Risko C, Chen CE, Breedas JL, McGehee MD, Sellinger A (2011) Chem Mater 23:5484
Shin RYC, Kietzke T, Sudhakar S, Dodabalapur A, Chen Z-K, Sellinger A (2007) Chem Mater 19:1892–1894
Sahika Inal MC, Sellinger A, Neher D (2009) Macromol Rapid Commun 30:1263–1268
Delley B (1990) J Chem Phys 92:508
Delley B (2000) J Chem Phys 113(18):7756-7764
Kresse G, Furthmüller J (1996) Comput Mater Sci 6:15
Wang Y, Perdew JP (1991) Phys Rev B 45(23):13244
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77(18):3865–3868
Becke AD (1988) J Chem Phys 88:2547
Becke AD (1993) J Chem Phys 98:5648–5652
Hehre WJ, Radom L, Schleyer PVR, Pople JA (1986) Ab initio molecular orbital theory. Wiley, New York
Rasmussen PG, Reybuck SE, Johnson DM, Lawton RG (2000) United States Patent-US006096899A
Johnson DM, Rasmussen PG (2000) Macromolecules 33(23):8597–8603
Fukui K, Yonezawa T, Shingu H (1952) J Chem Phys 20(8):722–725
Boese AD, Handy NC (2001) J Chem Phys 114(13):5497–5503
Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, Fiolhais C (1992) Phys Rev B 46(11):6671–6687
Vosko SH, Wilk L, Nusair M (1980) Can J Phys 58(8):1200–1211
Martin RM (2004) Electronic structure: basic theory and practical methods. Cambridge University Press, Cambridge
Gutiérrez-Pérez R-M, Flores-Holguín N, Glossmann-Mitnik D, Rodriguez-Valdez L-M (2011) J Mol Model 17:1963–1972
B. A. D (1993) J Chem Phys 98:5648–5647
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mohamad, M., Ahmed, R., Shaari, A. et al. First principles investigations of vinazene molecule and molecular crystal: a prospective candidate for organic photovoltaic applications. J Mol Model 21, 27 (2015). https://doi.org/10.1007/s00894-015-2582-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-015-2582-8