
Abstract
In this paper, coupled cluster methods CC2, CCSD, CCSDR(3) and EOM-CCSD(T) have been benchmarked against CC3 for the transition energies of nucleobases. Beside presenting vertical excitation energies for about 30 singlet transitions of four molecules, the results are analyzed statistically and problematic cases have been discussed in detail. It is concluded that the mean deviation of the CC2 results is smaller than that of the CCSD. However, the latter seems to be more systematic, i.e. it usually overestimates excitation energies by about 0.2 eV but with somewhat smaller standard deviation. Unfortunately, with decreasing single excitation contribution in the wave function CCSD gives large error, which can not be corrected by the non-iterative triples methods CCSDR(3) and EOM-CCSD(T).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lots of attention have been payed to the theoretical description of the excited states of nucleobases, for recent reviews see Ref. [1]. Of interest are vertical excitation energies, excited state potential energy surfaces, conical intersections, and finally, dynamics of the excitation process. The latter studies are very important for describing and understanding the details of processes which take place after nucleobases, the building blocks of DNA, are irradiated by UV light.
It has been shown recently by Barbatti et al. [2], that the details of the potential energy surface, location of conical intersections influence the outcome of such dynamics simulation by a great deal. Therefore it is extremely important to get accurate surfaces, and in particular the relative energy of different excited states need to be obtained correctly. Such studies for nucleobases, hydrated nucleobases, nucleotides, and dimers of nucleobases have been published recently by us [3–5].
Several methods can be used to study the excited states of nucleobases and these are extremely different in their approximations. Time Dependent DFT (TDDFT) seems to be a cost effective choice, which gives quite accurate results for most molecules [6]. However, in case of the building blocks of DNA, several problems arise, due to improper description of the large π-systems, charge transfer and Rydberg states [7, 8]. CASSCF and CASPT2, as flexible tools for describing excited states [9, 10], are also often applied. The disadvantage here is that only a very careful use of these methods (proper selection of the reference space, appropriate dumping factors) can result in reliable results [11, 12]. Coupled-Cluster (CC) methods [13, 14] may also be used for excited states. Equation of Motion (EOM) [15], or Linear Response (LR) [16, 17] versions of CC theory provide a hierarchical set of methods where quality can be controlled by the excitation level: EOM-CCSD [18] or CCSD-LR [19] is the lowest level including single and double excitations, one can go to higher level such as EOM-CCSDT [20], or even higher [21]. In addition, there exist methods in between with perturbation theory arguments simplifying the equations. Most popular is CC2 [22], which should be considered as an approximation to CCSD. Similar approach in EOM-style was also suggested (e.g. EOM-CCSD(2) [23], or Partitioned EOM-MBPT(2) [24]). There are also a series of methods including triples approximately on the top of the CCSD: as example one can name EOM-CCSDT-3 by Watts and Bartlett [25], CC3 [26].
In a recent paper [27] we have presented benchmark results obtained by Coupled-Cluster methods on over 100 singlet states of 28 molecules (test set of Thiel et al. [11]). The conclusion of that paper concerning the accuracy of the CC2-CCSD-CC3 series where somewhat different than the conclusion obtained in our earlier paper on the nucleobases [3]. Therefore we have reinvestigated the performance of these and similar methods on the nucleobases cytosine, adenine, thymine and uracil.
Methods
In this paper we apply the hierarchy CC2-CCSD-CC3 for the calculation of excitation energies of nucleobases. CC2 [22] is a second order version of CCSD, specifically designed for excited states within the linear response framework [17]. After some general papers on the subject [15, 16], the LR version of CCSD theory was put forward by Koch et al. [17, 19], while the Equation of Motion (EOM) version was coded and published shortly after by Bartlett and coworkers [18, 28]. These two methods are equivalent if excitation energy is of interest [14, 29], but differ for transition moment [29]. CC3 includes already approximate triples [26] in an iterative way, while CCSDR(3) [30] does it non-iteratively. Note that similar triples containing methods were derived earlier by Watts and Bartlett (EOM-CCSDT-1 [31] and EOM-CCSDT-3 [25], as well as EOM-CCSD(T) [25]) which were shown to give very similar results for a large set of excited states with the LR versions [32].
The calculations have been performed with different programs. CFOUR [33] has been used for CC2, CCSD, CC3 and EOM-CCSD(T) excitation energy calculations, Dalton2013 [34, 35] was used for LR type CC2, CCSD and CC3 excitation energies and oscillator strength, as well as CCSDR(3) excitation energies.
TZVP basis set [36] was used as in the calculation on the larger set of molecules [27]: since this basis does not include diffuse functions, Rydberg states are not handled properly. This introduces some artifacts into the results due to unphysical mixing of states, but does not prevent us to compare the results obtained by different methods. If necessary we will mention these complications in the discussion below. The core electrons have been frozen in all calculations presented here. Geometries have also been taken from Ref. [11], which are optimized structures at the MP2/6-31G ∗ level.
For the representation of the character of excited states the natural orbitals of the difference density of the ground and excited states were applied. These natural orbitals, along with the corresponding occupation numbers, allow an unbiased characterization of π−π ∗, n−π ∗ and also Rydberg transitions as has been demonstrated in our earlier papers for nucleobases [3, 37], nucleotides [4] and nucleobases dimers [5]. The single excitation contribution of an excited state is measured as in Dalton [34], i.e. by the norm of the EOM eigenvector in the singles space. The values given are obtained from the CC3 calculations.
Results and discussion
Table 1 shows the results of all calculations. Excitation energies and oscillator strengths are listed which were obtained at the CC2, CCSD, CC3, CCSDR(3) and EOM-CCSD(T) levels of theory. Oscillator strengths are given in the LR representation (see above). In addition, singles contribution to the excited state CC3 wave function is also listed, as calculated by Dalton [34].
Some notes are necessary for the assignment of the states. This is very important for a proper comparison of the performance of the different methods and for understanding their deficiencies. These assignments have been done by inspecting the natural orbitals of the difference density (CC2 and CCSD) as well as the leading coefficients of the expansion vectors.
There were no ambiguity assigning π−π ∗ transitions for any of the molecules. This means also that the order of different π−π ∗ transitions was found to be the same by the different methods. On the other hand, for the n−π ∗ transitions, we have discovered some complications in case of cytosine and uracil.
In Fig. 1 the natural orbitals of the difference density are given for the first four n−π ∗ transitions of cytosine in case of CC2 and CCSD wave functions. There are two lone pair sites in cytosine: the oxo group and that of the nitrogen in the ring. Excitations from these to two of the π ∗ orbitals (one involving the carbonyl, the other the ring π system and the NH2 group) results in four low lying transitions shown in the figure. The first and the third are mainly excitations from the oxygen lone pair, while the second and fourth are excitations mainly from the nitrogen lone pair. In case of CCSD the mixing is a bit smaller than in case of CC2. Concerning the π ∗ orbitals, the first two excitations are to the ring π ∗ orbital, the other two involve also the carbonyl group. In the second and fourth cases the CC2 and CCSD orbitals are hardly distinguishable. For the first and third, on the other hand, CC2 involves either C-NH2 or carbonyl anti-bonding contributions, in case of CCSD both contribute to both orbitals. Clearly, the form of the excitations are different in case of CC2 and CCSD. In case of CC3 the natural orbitals are not available, the coefficients of the EOM expansion vectors
show that the CC2 vectors are much more similar to CC3 one than to CCSD one: the dominant configuration(s) are always the same in the former case. This is not the case for CCSD: the vector of the second transition is a strong combination of the second and fourth vectors of CC3.

Natural orbitals of the difference density representing the lowest four n−π ∗ excitations of cytosine as obtained at the CC2 and CCSD levels. The orbitals are given in pairs, the left one has negative eigenvalue (hole orbitals, where the electron excited from), while the right one has positive eigenvalue (particle orbital, where the electron is excited to)
This is in line with the error of the excitation energies: while CC2 is close to CC3 (note however, that discrepancy is negative and somewhat larger for the first two states), in case of CCSD, the second and third n−π ∗ transitions are overestimated by 0.48 and 0.40 eV, respectively. This is 0.1-0.2 eV larger discrepancy than the average value found for the whole set [27] (see also below). While the larger error is clearly in line with the different mixing of basic excitation components, this analysis does not give an explanation for different performance of CC2 and CCSD.
In Fig. 2 the same natural orbitals are shown for uracil. In this case there are two oxo lone pairs and two carbonyl π ∗ orbitals. For the first two n−π ∗ transitions, the orbitals obtained from the CC2 and CCSD wave functions are very similar, and they can be assigned as transitions from one of the oxygen lone pairs to the corresponding carbonyl π ∗ orbital. In case of the other two transitions, differences can be observed. The two most important detachment orbitals of the 4 \(^{1}\textit {A}^{\prime \prime }\) transition obtained from the CC2 density are mostly localized on each oxygen atoms, in case of CCSD we observe two combinations of the individual lone pairs. The major component of the attachment orbital is the same in the two cases, the orbital with the second largest occupation number is an almost pure oxygen lone pair (combination on the two oxygen atoms) in case of CCSD, while in case of CC2 it resembles the attachment orbital of the 2 \(^{1}\textit {A}^{\prime \prime }\) transition, i.e. has some ring π ∗ component. The detachment orbitals of the 5 \(^{1}\textit {A}^{\prime \prime }\) transition, in case of CCSD, are very similar to the ones of 4 \(^{1}\textit {A}^{\prime \prime }\). The same orbitals appear in case of CC2, but with somewhat different occupation numbers. The most important attachment orbital is a combination of carbonyl π ∗ orbitals both in CCSD and CC2 cases. While this is dominant in case of CC2, in the CCSD case there is an almost pure ring π ∗ orbital with occupation number of 0.33. In summary, the character of the first two n−π ∗ transitions obtained at the CC2 and CCSD levels are the same and we observed slight differences in case of the third and fourth transitions, the contribution from the ring π ∗ orbital seems to be reversed between 4 \(^{1}\textit {A}^{\prime \prime }\) and 5 \(^{1}\textit {A}^{\prime \prime }\) transitions in CC2 and CCSD cases. Considering finally the dominant contributions of the expansion vectors, again CC2 and CC3 seem to agree, while there is different mixing in case of the second and fourth transitions for CCSD.

Natural orbitals of the difference density representing the lowest four n−π ∗ excitations of uracil as obtained at he CC2 and CCSD levels. The orbitals are given in pairs, the left one has negative eigenvalue (hole orbitals, where the electron excited from), while the right one has positive eigenvalue (particle orbital, where the electron is excited to)
Very large discrepancy could be found for CCSD excitation energies of uracil for the third and fourth n−π ∗ transitions. Although we see some differences between the character of these transitions in case of CC2 and CCSD, this does not provide an immediate explanation for the failure of CCSD. Later we come back to this problem and try to explain it from an other point of view.
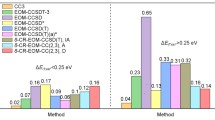
In Table 2 statistical analysis of the CCSD and CC2 results is presented, separate columns are given for the π−π ∗ and n−π ∗ transitions (the column “all” also includes the states assigned as Rydberg in Table 1). The corresponding graphical representation is shown in Fig. 3.

Deviation of excitation energy (eV) obtained at the CC2, CCSD, CCSDR(3) and CCSD(T) levels with respect to the CC3
For the first glance, the statistics for these states are similar to that for the large set in Ref. [27]. Note that practically all states have single excitation contribution between 80 and 90 %. There are two facts to recognize. First, for CC2 the mean value for the n−π ∗ states is -0.03 eV, while for the π−π ∗ states it is 0.09 eV. This is in line with the conclusion of Ref. [3]: CC2 seems to underestimate the n−π ∗ excitation energies of the nucleobases systematically. Since, on the other hand, the π−π ∗ excitations are overestimated, the energy difference of these states are presumably not very well predicted by the CC2 method. In case of CCSD the mean error is a bit larger than before (0.3 eV), but it is practically the same for both types of states.
The second important observation is that the maximum error in case of CCSD is quite large. This is caused by n−π ∗ transitions of uracil (4 \(^{1}\textit {A}^{\prime \prime }\) and 5 \(^{1}\textit {A}^{\prime }\)) and cytosine (2 \(^{1}\textit {A}^{\prime \prime }\)), as discussed above already. In case of the π−π ∗ transitions the largest discrepancy has also been found for uracil (3 1 A ′ transition) and for thymine which is structurally similar. Note that, except one of these, the corresponding transition energies are above 6 eV, therefore less important from the spectroscopy point of view. For all other transitions the discrepancy is much smaller (0.34 eV) and in line with the value found for the larger set [27].
Above we failed to explain the large discrepancy of CCSD excitation energies in case of uracil and cytosine. With Fig. 4 we do one more attempt: here we plot the error of CC2 and CCSD with respect to CC3 as a function of the singles contribution in CC3 wave function. It is seen that in case of CCSD for both n−π ∗ and π−π ∗ transitions the largest errors appear with decreasing singles contribution. This is a well know general property of EOM (or LR) methods at the SD level: these can only be applied for transitions with dominantly single excitation character. The figure shows clearly that for both types of excitations the error gets large when the singles contribution drops below 84-85 % and therefore these values seem to be a limit for accurate applications. Surprisingly, CC2 does not seem to deteriorate that much with the decreasing singles contribution, however, a significant difference of the mean error in case of the n−π ∗ and π−π ∗ transitions can be clearly seen on the figure. According to this figure, CCSD performs worse, however, by leaving out the five worst cases (discussed above), the rest of the points are quite systematic and the errors do not differ a lot for the two types of excitations.

Dependence of the error of the CC2 (left) and CCSD (right) excitation energies as a function of the singles contribution of the EOM vector. The measure of singles contributions is from CC3 calculations
In Ref. [3] we have suggested that in case of nucleobases CC2 often gives the wrong order of the π−π ∗ and n−π ∗ states and no such case have been detected for CCSD. The left panel of Fig. 4 shows clearly that CC2 systematically underestimates n−π ∗ and overestimates π−π ∗ transition energies. Closer investigation of Table 1 shows, however, that in some cases also CCSD predicts different order of states with respect to CC3. The mixed-up order of states with respect to CC3 appearing in Table 1 can be summarized as follows:
-
3 \(^{1}\textit {A}^{\prime }\) and 2 \(^{1}\textit {A}^{\prime \prime }\) states of cytosine in case of CCSD. Note that here the two states are within 0.1 eV at the CC3 level, while the CCSD energy difference is smaller than 0.05 eV.
-
3 \(^{1}\textit {A}^{\prime }\) and 2 \(^{1}\textit {A}^{\prime \prime }\) states of thymine for both CC2 and CCSD. In case of CCSD 3 \(^{1}\textit {A}^{\prime }\) is one of the worst case among the π−π ∗ transitions with deviation of 0.44 eV from CC3. In case of CC2, indeed the error of n−π ∗ and π−π ∗ have opposite sign.
-
3 \(^{1}\textit {A}^{\prime }\) and 2 \(^{1}\textit {A}^{\prime \prime }\) states of uracil for again both for CC2 and CCSD. Explanation is the same as for thymine (see also the discussion in the previous paragraph).
-
2 \(^{1}\textit {A}^{\prime }\) and 2 \(^{1}\textit {A}^{\prime \prime }\) states of adenine in case of CC2, but in this case CC3 transition energies are also almost degenerate.
From these observations one can conclude that mixup of states of different types can appear for both CCSD and CC2 and can have two causes: i) very small energy gap between the two types of states and ii) large error of one of the states as appeared in case of CCSD for uracil and thymine.
In Table 3 the statistical analysis for the non-iterative triples methods are presented. The mean deviation from the CC3 results are below 0.1 eV for both methods and transition types. Similarly small are the mean absolute deviations showing great deal of consistency. Maximum deviations are, however, quite large: for the 2 \(^{1}\textit {A}^{\prime \prime }\) state of cytosine it is 0.32 eV in case of CCSDR(3) and 0.34 eV in case of CCSD(T). Note that this is exactly the state where we have found one of the largest error in case of CCSD. It seems that non-iterative triples corrections do not work in this case. Similarly, the CCSDR(3) and CCSD(T) discrepancies are 0.28 eV and 0.34 eV, respectively, for the 4 \(^{1}\textit {A}^{\prime \prime }\) state of uracil which again was discussed as a problematic case for CCSD. Both mentioned states of cytosine and uracil have a single excitation contributions below 84 % which appears to be a limit for accurate application here again.
Although CC2 is at much lower level in the hierarchy of CC theory than non-iterative triples methods, the mean deviation and the RMS are smaller for CC2 than for the more expensive methods. As this is most probably the result of error cancellation, the mean absolute deviation and the standard deviation is smaller for the triples methods showing that these give more systematic results. Still, in same cases, when CCSD fails badly, the non-iterative methods tend to do so, too. This is the consequence of the fact that the single and double part of the wave function is not changed by the triples correction in these non-iterative methods.
Conclusions
In this paper CC2, CCSD, CCSDR(3) and EOM-CCSD(T) methods have been benchmarked against the CC3 method for the singlet excitation energies of nucleobases. The latter technique was proven quite accurate (error smaller than 0.1 eV) compared to EOM-CCSDT for smaller molecules [27].
As for a larger set of 28 molecules and about 150 singlet excitetd states in Ref. [27], now also for nucleobases one can conclude that both CC2 and CCSD gives results as accurate as 0.1-0.3 eV in most cases. Statistical analysis shows that CC2 predicts the excitation energies with a mean deviation close to zero, while CCSD systematically overestimates these by about 0.2-0.3 eV. Disadvantage for CCSD is that with the single excitation contribution decreasing below a certain level (84-85 % was diagnosed here), it can eventually fail badly. This was less of a problem in case of the larger set, nucleobases seem to be a special case in this respect. Even by carefully analyzing the wave function, we could not find a reasonable explanation for this failure. Advantage of CC2 in this respect is that the results deteriorate with decreasing single excitation contribution much slower, in fact the results remain quite systematic for all states investigated in this paper. On the other hand, a disadvantage of CC2 is that the excitation energies are above CC3 values for π−π ∗ transitions, while in case of n−π ∗ transitions the trend is opposite. Excluding the five very bad values, CCSD is more systematic, the average error of the two types of transition is the same.
Non-iterative triples corrections (CCSDR(3) and EOM-CCSD(T)) improve the results in all cases. Statistically the results are better than CCSD (smaller average error) and more systematic than CC2 (smaller deviation from average). However, none of these methods can cure the excitation energies completely in those cases where the underlying CCSD fails.
As final conclusion, in contrary to what has been said in Ref. [3], CC2 seems to be a good tool to obtain the vertical excitation energy of nucleobases. Two questions remain open: first, it has not been investigated how much the conclusions of this paper are basis set dependent, in particular, diffuse functions may affect the accuracy of the methods. The second question is whether the present conclusion is valid also for the potential energy surfaces, or the conclusion is a consequence of the common geometry used to obtain vertical excitation energies. These questions will be investigated in forthcoming papers.
References
Kleinermanns K, Nachtigallova D, de Vries MS (2013). Int Rev Phys Chem 32:308
Barbatti M, Lan Z, Crespo-Otero R, Szymczak J J, Lischka H, Thiel W (2012). J Chem Phys 137:22A503
Szalay P G, Watson T J Jr, Perera A, Lotrich V F, Bartlett R J (2012). J Phys Chem A 116:6702
Szalay P G, Watson T J, Watson T J Jr, Perera A, Lotrich V F, Fogarasi G, Bartlett R J (2012). J Phys Chem A 116:8851
Szalay P G, Watson T J Jr, Perera A, Lotrich V F, Bartlett R J (2013). J Phys Chem A 117:3149
Dreuw A, Head-Gordon M (2005). Chem Rev 105:4009
Lange A W, Rohrdanz M A, Herbert J M (2008). J Phys Chem B 112:6304
Lange A W, Herbert J M (2007). J Chem Theory Comput 3:1680
Andersson K, Malmqvist P-A A, Roos B O, Sadlej A J, Wolinski K (1990). J Chem Phys 94:5483
Andersson K, Malmqvist P-A A, Roos B O (1992). J Chem Phys 96:1218
Schreiber M, Silva M R J, Sauer S P A, Thiel W (2008). J Chem Phys 128:134110
Szalay P G, Mueller T, Gidofalvi G, Lischka H, Shepard R (2012). Chem Rev 112:108
Bartlett R J, Musial M (2007). Rev Mod Phys 79:291
Sneskov K, Christiansen O (2011). WIREs Comput Mol Sci 2:566
Sekino H, Bartlett R J (1984). Int J Quantum Chem 255
Monkhorst H J (1977). Int J Quantum Chem 421
Koch H, Jørgensen P (1990). J Chem Phys 93:3333
Stanton J F, Bartlett R J (1993). J Chem Phys 98:7029
Koch H, Jensen H J A, Jørgensen P, Helgaker T (1990). J Chem Phys 93:3345
Kucharski S A, Wloch M, Musial M, Bartlett R J (2001). J Chem Phys 115:8263
Kállay M, Gauss J (2004). J Chem Phys 121:9257
Christiansen O, Koch H, Jørgensen P (1995). Chem Phys Lett 243:409
Stanton J F, Gauss J (1995). J Chem Phys 103:1064
Gwaltney S R, Bartlett R J (1999). J Chem Phys 110:62
Watts J D, Bartlett R J (1996). Chem Phys Lett 258:581
Christiansen O, Koch H, Jørgensen P (1995). J Chem Phys 103:7429
Kánnár D, Szalay P. G (2014). J Chem Theory Comput 10:3757
Comeau D C, Bartlett R J (1993). Chem Phys Lett 207:414
Koch H, Kobayashi R, Sanchez de Merás A, Jørgensen P (1994). J Chem Phys 100:4393
Christiansen O, Koch H, Jørgensen P (1996). J Chem Phys 105:1451
Watts J D, Bartlett R J (1995). Chem Phys Lett 233:81
Watson T J Jr, Lotrich V F, Szalay P G, Perera A, Bartlett R J (2013). J Phys Chem A 117:2569
CFOUR, a quantum chemical program package written by Stanton JF, Gauss J, Harding ME, Szalay PG with contributions from Auer AA, Bartlett RJ, Benedikt U, Berger C, Bernholdt DE, Christiansen O, Heckert M, Heun O, Huber C, Jonsson D, Jusélius J, Klein K, Lauderdale WJ, Matthews D, Metzroth T, O’Neill DP, Price DR, Prochnow E, Puzzarini C, Ruud K, Schiffmann F, Schwalbach W, Simmons C, Stopkowicz S, Tajti A, Vázquez J, Wang F, Watts JD and the integral packages MOLECULE (Almlöf J and Taylor PR), PROPS (Taylor PR), ABACUS (Helgaker T, Jensen HJ Aa, Jørgensen P, and Olsen J), and ECP routines by Mitin A V and van Wüllen C. For the current version, see http://www.cfour.de.
Dalton, a molecular electronic structure program, release Dalton2013.2 (2013), see http://daltonprogram.org
Aidas K, Angeli C, Bak K L, Bakken V, Bast R, Boman L, Christiansen O, Cimiraglia R, Coriani S, Dahle P, Dalskov E K, Ekström U, Enevoldsen T, Eriksen J J, Ettenhuber P, Fernández B, Ferrighi L, Fliegl H, Frediani L, Hald K, Halkier A, Hättig C, Heiberg H, Helgaker T, Hennum A C, Hettema H, Hjertenaes E, Høst S, Høyvik I-M, Iozzi M F, Jansík B, Jensen H J A, Jonsson D, Jørgensen P, Kauczor J, Kirpekar S, Kjaergaard T, Klopper W, Knecht S, Kobayashi R, Koch H, Kongsted J, Krapp A, Kristensen K, Ligabue A, Lutnaes O B, Melo J I, Mikkelsen K V, Myhre R H, Neiss C, Nielsen C B, Norman P, Olsen J, Olsen J M H, Osted A, Packer M J, Pawlowski F, Pedersen T B, Provasi P F , Reine S, Rinkevicius Z, Ruden T A, Ruud K, Rybkin V V, Sałek P, Samson C C M, de Merás A S, Saue T, Sauer S P A, Schimmelpfennig B, Sneskov K, Steindal A H, Sylvester-Hvid K O, Taylor P R, Teale A M, Tellgren E I, Tew D P, Thorvaldsen A J, Thøgersen L, Vahtras O, Watson M A, Wilson D J D, Ziolkowski M, Ågren H (2013). WIREs Comput Mol Sci 4:269
Schäfer A, Horn H, Ahlrichs R (1992). J Chem Phys 97:2571
Benda Z, Szalay P G (2014). J Phys Chem A 118:6197
Acknowledgments
This work has been supported by Orszagos Tudomanyos Kutatasi Alap (OTKA; Grant No. 104672). The authors greatfully thank to John F. Stanton for helping with the implementation of the EOM-CCSD(T) method into the CFOUR program.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper belongs to Topical Collection Brazilian Symposium of Theoretical Chemistry (SBQT2013)
The authors acknowledge support by OTKA (grant number F104672).
Rights and permissions
About this article

Cite this article
Kánnár, D., Szalay, P.G. Benchmarking coupled cluster methods on singlet excited states of nucleobases. J Mol Model 20, 2503 (2014). https://doi.org/10.1007/s00894-014-2503-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-014-2503-2