
Abstract
The adsorption of molecular hydrogen on few-layer graphene (FLG) structures is studied using molecular dynamics simulations. The interaction between graphene and hydrogen molecules is described by the Lennard-Jones potential. The effects of pressure, temperature, number of layers in a FLG, and FLG interlayer spacing are evaluated in terms of molecular trajectories, binding energy, binding force, and gravimetric hydrogen storage capacity (HSC). The simulation results show that the effects of temperature and pressure can offset each other to improve HSC. An insufficient interlayer spacing (0.35 nm) largely limits the HSC of FLG because hydrogen adsorbed at the edges of the graphene prevents more hydrogen from entering the structure. A low temperature (77 K), a high pressure, a large number of layers in a FLG, and a large FLG interlayer spacing maximize the HSC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hydrogen is a potential next-generation clean fuel for replacing fossil fuels. It can reduce CO2 emissions and air pollution due to its high power of combustion, clean combustion, and renewability. Hydrogen-based fuel cells [1] have been applied to efficiently produce electricity for mobile applications. A critical requirement is hydrogen storage systems that can operate at ambient conditions with sufficient gravimetric and volumetric capacities.
Carbon-based nanomaterials (i.e., carbon nanotubes (CNTs) and fullerences) that comprise carbon atoms with sp2 hybridization are relatively light and inexpensive, and have a high adsorption surface area for hydrogen storage. Graphene, which consists of single-atom-thick sheets of carbon, is the basic structural element of these carbon nanomaterials. Graphene has excellent mechanical (Young’s modulus ∼1.0 TPa), thermal (thermal conductivity ∼3000 W/mK), and electronic properties [2–4]. Several theoretical studies on graphene have demonstrated that it can absorb up to an 8∼9 % mass ratio of hydrogen, which is close to the desired gravimetric capacity set by the US Department of Energy for hydrogen storage (9 wt% by 2015).
Understanding the mechanisms of hydrogen adsorption on graphene under various environments would benefit various fields, including motor vehicles, fusion reactor design, and hydrogen storage [5]. Molecular dynamics (MD) simulations of a graphene–hydrogen interface can be used to explore the nature of surface adsorption. Atomistic simulation can effectively avoid experimental noise and turbulence problems. Many nanosystems have been analyzed using MD, such as metal films [6], nanowires [7, 8], nanoimprinting [9], and dip-pen nanolithography [10, 11]. Herrero and Ramirez [12] studied the diffusion of hydrogen in graphite and found that hydrogen atoms jump from a C atom to a neighboring one with an activation energy of about 0.4 eV. Compared to the adsorption of atomic hydrogen on graphene, molecular hydrogen has a much smaller activation energy and faster diffusion speed due to the interaction of physisorption [13, 14] with graphene. Lamari and Levesque [15] studied hydrogen adsorption on graphene and found that at a temperature of 77 K and a pressure of 1 MPa, the excess hydrogen physisorption is estimated to be equal to ∼7 wt%, and decreases with increasing temperature. Tozzini and Pellegrini [16] studied a reversible hydrogen storage process on graphene utilizing density-functional theory. The reversible storage and release of hydrogen are controlled by the extent of graphene buckling, which leads to a high gravimetric capacity of up to 8 wt%. Graphane is a fully saturated hydrocarbon compound derived from a single graphene sheet with formula CH, which has a high gravimetric capacity of 7.7 wt% with great stability [17].
This work investigates the mechanism of molecular hydrogen adsorption and gravimetric capacity on a few-layer graphene (FLG) structure using MD simulations. To optimize the operation parameters, the effects of pressure, temperature, number of layers in a FLG, and FLG interlayer spacing are studied. The results are discussed in terms of molecular trajectories, binding energy, binding force, and gravimetric hydrogen storage capacity (HSC).
Methodology
Figure 1 (a) shows an initial physical model of a hydrogen/graphene interface. The single-layer graphene comprises C atoms and is arranged in the form of a honeycomb lattice. Hydrogen molecules with a face-centered cubic (fcc) structure are placed above and below the graphene. The hydrogen molecules are treated as single spherical molecules to simplify the model. The graphene sheet has dimensions of 5 (length) × 5 nm (width). There are 4000 hydrogen molecules in the simulated system, whose dimensions are 7 (length) × 7 (width) × 10 nm (height), corresponding to a gas density of 1.4 g/m3. Periodic boundary conditions are applied in all three dimensions. To focus on the hydrogen adsorption mechanism, the graphene is considered to be ideally rigid to increase computational efficiency.

Initial MD physical model of a hydrogen/single-layer graphene interface
The computer simulation codes incorporating Hamiltonian dynamics were developed in-house by the authors to simulate the hydrogen adsorption. The systems were maintained at constant temperatures and pressures by the velocity and volume rescalings [18], respectively. The isothermal-isobaric (NPT) ensemble is used in the system. The Lennard-Jones potential [19] is adapted to describe the van der Waals (VDW) interactions of C/H2 and H2/H2. The potential parameters are listed in Table 1. A cut-off radius of 1.0 nm is used to evaluate the number of hydrogen molecules adsorbed on the graphene. The time step is fixed at 1 fs. The pressure, temperature, FLG interlayer spacing, number of layers in a FLG are set in the ranges of 6 to 15 MPa, 77 to 400 K, 0.35 to 1.40 nm, and 1 to 4 layers, respectively, to determine their effects on HSC. The hydrogen interacting with the FLG begins after all hydrogen underwent a MD equilibrium run of 100 ps in order to achieve thermal equilibrium and a uniform distribution in the system.
Results and discussion
Effects of pressure and temperature on HSC of single-layer graphene
Pressure and temperature are the two most significant parameters for the control of gas storage in addition to material selection. The effects of pressure and temperature on the hydrogen adsorption on single-layer graphene are individually discussed. Pressure values of 6, 8, 10, and 15 MPa are used in the system to investigate the pressure effect. The system temperature is maintained at 77 K. Figures 2 (a) and (b) show variations of binding energy and binding force with time for pressures of 6 to 15 MPa, respectively. The binding energy and binding force are calculated by summing the interactive energy and force from the graphene interacting with hydrogen, respectively. Note that both binding energy and binding force are negative values and decrease with increasing number of hydrogen molecules adsorbed onto the surface. In Fig. 2 (a) and (b), in the first 150 ps, the energy and force curves rise with time. This is due to the instant repulsive interactions between C and H2 due to an overlap, indicating an unstable adsorption period. Both curves then significantly decay with increasing time, which indicates that the amount of adsorbed hydrogen gradually increases. The result shows that an increase in system pressure leads to a lower (negative) binding energy and binding force. The binding energy and binding force converge after 1500 ps. Figure 3 (a) shows the variation of gravimetric hydrogen with time for these pressures. Gravimetric hydrogen is defined as the mass ratio of hydrogen molecules adsorbed on the graphene to all atoms in the system. According to the curves, the hydrogen adsorption process can be divided into three periods. At the time period of 0∼325 ps, gravimetric hydrogen increases. A more rapid increase appears in the following time period (325∼910 ps). Then, the increase becomes slow, indicating a gradual convergence (close to a saturate state). After 1500 ps, two to three molecular layers of hydrogen naturally form on the graphene surface due to the VDW interaction. A larger density of hydrogen layers on graphene can be obtained by increasing system pressure, as shown in snapshots of Fig. 3 (a). The gravimetric hydrogen increases 1.29-, 1.45-, and 1.73-fold when the system pressure is increased from 6 to 8, 10, and 15 MPa, respectively.

Variations of (a) binding energy and (b) binding force with time at a temperature of 77 K for pressures of 6∼15 MPa

Variation of gravimetric hydrogen with time at (a) a temperature of 77 K for pressures of 6∼15 MPa and (b) a pressure of 10 MPa for temperatures of 100∼400 MPa
The effect of temperature is studied using temperatures of 100, 200, 300, and 400 K. The system pressure is fixed at 10 MPa. Figure 3 (b) shows the variation of gravimetric hydrogen with time for these temperatures. The gravimetric hydrogen largely decreases with increasing temperature due to an increase in the kinetic energy of hydrogen molecules. Due to a weak C-H2 binding energy (approximately units of kcal mol−1) [20], the hydrogen molecules adsorbed on graphene are easily desorbed when their kinetic energies increase with temperature, which is in agreement with previous reports [15, 21]. At stage I, high-speed adsorption appears. The gravimetric hydrogen then gradually slows down with time (stages II and III). The gravimetric hydrogen obtained at 77 K (Fig. 3(a) and (b)) is higher than that obtained at higher temperatures. The gravimetric hydrogen increases 1.27-, 1.45-, and 1.71-fold when the system temperature is decreased from 400 K to 300, 200, and 100 K, respectively. The results indicate that HSC has a strong dependence on pressure and temperature. The effects of the two parameters are expected to partially offset each other to improve HSC. HSC is optimal in a system with a high pressure and a low temperature.
Effect of number of layers in a FLG
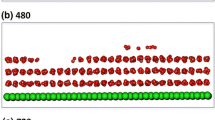
FLG is considered better than single-layer graphene as a hydrogen storage material due to its larger adsorbable area. FLG consists of multiple weak interlayer interfaces which are combined by VDW interaction. An interlayer spacing of 0.35 nm in a FLG was obtained through both experiment [22] and theory [23]. However, in the present study, the simulation of FLG-hydrogen adsorption reveals that hydrogen cannot easily enter the FLG structure due to the insufficient interlayer spacing, and thus the hydrogen molecules adsorb only at the outer surface. This indicates that the efficiency of hydrogen storage for a FLG might be improved by suitably adjusting the interlayer spacing. Figure 4 shows the variation of gravimetric hydrogen with the number of layers in a FLG at a temperature of 77 K and a pressure of 10 MPa. An interlayer spacing of 0.70 nm is used. When the interlayer spacing is increased from 0.35 to 0.70 nm, a few hydrogen molecules flow into structure and are adsorbed inside, as shown in the snapshots of Fig. 4. The gravimetric hydrogen significantly increases with the number of layers in a FLG due to an increase in the internal storage space. It increases 4.06, 6.91, and 11.15 wt% when the number of layers is increased from 1 to 2, 3, and 4, respectively. The results show that increasing the number of layers in a FLG can significantly improve the amount of gravimetric hydrogen under a sufficient interlayer spacing.

Variation of gravimetric hydrogen with number of layers in a FLG for an interlayer spacing of 0.70 nm at a temperature of 77 K and a pressure of 10 MPa
Effect of interlayer spacing
This section focuses on the effect of FLG interlayer spacing on hydrogen storage. Interlayer spacings of 0.35, 0.70, 1.05, and 1.40 nm are used. The FLG is set to four layers. Figure 5 shows snapshots of hydrogen adsorption and the variation of gravimetric hydrogen at a temperature of 77 K and a pressure of 10 MPa for various interlayer spacings. The lowest hydrogen storage appears at an interlayer spacing of 0.35 nm (original), which is insufficient. The hydrogen molecules only adsorbed at the outer surfaces and around the edges of the FLG. With an interlayer spacing of 0.70 nm, a few hydrogen molecules flow into the interlayer spaces from the edges and gather inside through VDW interaction. At an interlayer spacing of 1.05 nm, more hydrogen molecules are adsorbed inside. The hydrogen molecules stored in the FLG form one to two layers of molecules adsorbed on the inner surfaces at an interlayer spacing of 1.40 nm. The gravimetric hydrogen increases 3.48-, 4.00-, and 4.21-fold when the interlayer spacing is increased from 0.35 to 0.70, 1.05, and 1.40 nm, respectively. The results indicate that gravimetric hydrogen can be effectively increased by increasing the FLG interlayer spacing, which significantly increases the adsorbable area and storage space. For the four-layers graphene, the saturated gravimetric hydrogen of about 9.6 wt% appears at an interlayer spacing of 1.60 nm. However, the amount of gravimetric hydrogen still slightly increases with further increasing interlayer spacing due to an increase in internal storage space instead of adsorbable area.

Variation of gravimetric hydrogen with interlayer spacing for four-layer graphene at a temperature of 77 K and a pressure of 10 MPa
Effects of pressure and temperature on HSC of FLG
The effects of pressure and temperature on the HSC of FLG are studied using a physical model of four-layer graphene. A large interlayer spacing of 1.40 nm is used to study the HSC of FLG. Figure 6 shows the relationship between gravimetric hydrogen and pressure (6 to 15 MPa) at a temperature of 77 K. When the pressure is increased from 6 MPa, the gravimetric hydrogen only slightly increases because the surfaces and internal structure of the graphene are almost filled with hydrogen (near saturation). The gravimetric hydrogen increases 0.33-, 0.53-, and 1.32-fold when the pressure is increased from 6 to 8, 10, and 15 MPa, respectively. Figure 7 shows the relationship between gravimetric hydrogen and temperature (77 to 300 K) at a pressure of 10 MPa. The hydrogen adsorbed on the graphene becomes unstable when the temperature is increased from 77 to 150 K. This is due to the kinetic energy of hydrogen being beyond its physisorption energy on graphene. Less hydrogen can stay in the graphene when the temperature is increased from 77 K. The theoretical HSC obtained at room temperature is close to an experimental HSC of 0.9 wt% [24]. The gravimetric hydrogen decreases 6.70-, 8.09-, and 8.45-fold when the temperature is decreased from 77 to 150, 230, and 300 K, respectively. The results show a strong dependence of HSC on temperature.

Variation of gravimetric hydrogen with pressure for four-layer graphene with an interlayer spacing of 1.40 nm at a temperature of 77 K

Variation of gravimetric hydrogen with temperature for four-layer graphene with an interlayer spacing of 1.40 nm at a pressure of 10 MPa
Comparison of various carbon-based systems for hydrogen storage
Hydrogen storage on various bare carbon-based systems has been studied theoretically and experimentally. The HSCs of single-walled carbon nanotubes (SWCNTs) obtained by experiments are about 4 wt% (at 0.1 MPa and 77 K) [25] and 1 wt% (at 0.1 MPa and 295 K) [26]. For double-walled carbon nanotubes (MWCNTs), the HSCs are close to 5 wt% (at 10 MPa and 300 K) [27] and 0.25 wt% (at 0.1 MPa and 300 K) [28]. The HSC of MWCNTs depends on their size of shells and number of shells. Studies have shown that the HSC of SWCNTs is better than that of MWCNTs. Graphite nanofibers are layered graphite nanostructures which have high HSCs of 6.5-10 wt% (at 12 MPa and 300 K) [29, 30]. The HSC of graphene is 0.9 wt% (at 10 MPa and 298 K) [24]. These results reveal that for carbon-based systems, higher HSCs are obtained at higher pressures and lower temperatures, and for systems with a larger adsorbable surface area. However, for a given carbon-based system, the theoretical HSCs are much larger than those obtained by experiments due to the assumption of an ideal crystalline material and an ideal environment. For example, SWCNTs and graphene have theoretical HSCs of 11.2 wt% (at 10 MPa and 77 K) [31] and 7.0 wt% (at 1 MPa and 77 K) [15], respectively. The maximum (theoretical) HSC might thus be achieved by properly designing the material and controlling the environment.
Some metal decorated systems are attractive for hydrogen storage materials at room temperature. They have higher hydrogen wt% capacity than bare carbon materials due to a larger bond strength interacting with hydrogen and hydrogen absorption ability. Transition metal-ethylene complexes formed by laser ablation exhibit a high HSCs of 12.0 wt% (at 0.1 MPa and 298 K) [32]. The HSCs of multi-functionalized naphthalene with Ti and Li metal atoms are 6.72 and 3.73 wt% (at 0.1 MPa and 298 K), respectively [33]. Nanoscale titanium-benzene complexes have a HSC of 6.0 wt% (at 0.1 MPa and 298 K) [34]. The HSC of C2H2Ti and C2H2Li complexes are 12.0 and 19.65 wt% (at 0.1 MPa and 300 K) [35], respectively. The HSC of C2H4V and C2H4V+ organometallic compounds are 11.32 and 13.28 wt% (at 0.1 MPa and 298 K) [36], respectively. The HSCs of Ti-acetylene (C2H2Ti) and Li-acetylene (C2H2Li) complex are 12.0 and 19.65 wt% (at 0.1 MPa and 298 K) [37], respectively.
Conclusions
MD simulation was used to investigate hydrogen adsorption on a FLG structure. The effects of pressure, temperature, number of graphene layers, and FLG interlayer spacing on HSC were analyzed. For the effect of temperature, high-speed adsorption appears in the first 325 ps, whereas for the effect of pressure, it appears in the time period of 325∼910 ps. The effects of temperature and pressure can offset each other to improve HSC. The hydrogen adsorbed on graphene is sensitive to temperature due to a weak C-H2 binding energy. The hydrogen molecules adsorbed on graphene are easily desorbed when their kinetic energies increase with temperature. The HSC of FLG can be improved by increasing the FLG interlayer spacing, which significantly increases the adsorbable area and storage space. A low temperature (77 K), a high pressure, a large interlayer spacing, and many layers maximize the HSC of FLG.
References
Hwang JJ (2012) Review on development and demonstration of hydrogen fuel cell scooters. Renew Sustain Energy Rev 16(6):3803–3815
Lee C, Wei X, Kysar W, Hone J (2008) Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 321:385–388
Castro Neto AH, Guinea F, Peres NMR, Novoselov KS, Geim AK (2009) The electronic properties of graphene. Rev Mod Phys 81:109–162
Stankovich S, Dikin DA, Dommett GHB, Kohlhaas KM, Zimney EJ, Stach EA, Piner RD, Nguyen ST, Ruoff RS (2006) Graphene-based composite materials. Nature 442:282–286
Schlapbach L, Zuttel A (2001) Hydrogen-storage materials for mobile applications. Nature 414:353–358
Wu CD, Fang TH, Lin JF (2012) Atomic-scale simulations of material behaviors and tribology properties for FCC and BCC metal films. Mater Lett 80:59–62
Sung PH, Wu CD, Fang TH, Weng CI (2012) Size effect on shape recovery and induced strain of NiTi nanowires. Appl Surf Sci 258(18):7064–7069
Sung PH, Wu CD, Fang TH (2012) Effects of temperature, loading rate, and nanowire length on torsional deformation and mechanical properties of aluminum nanowires investigated using molecular dynamics simulation. J Phys D: Appl Phys 45:215303-1-8 doi:10.1088/0022-3727/45/21/215303
Hsu QC, Lin YT, Chou DC, Wu CD (2012) Study on nanoimprint formability considered anti-adhesion layer for the (CH2)n polymer material by molecular dynamics simulation. Curr Nanosci 8(3):424–431
Wu CD, Fang TH, Wu TT (2012) Effects of humidity and temperature on laser-assisted dip-pen nanolithography array studied using molecular dynamics simulations. J Colloid Interface Sci 372(1):170–175
Wu CD, Fang TH, Lin JF (2012) Effects of tip gap, deposition temperature, holding time, and pull-off velocity on dip-pen lithography using molecular dynamics simulation. J Appl Phys 111 (10):103521-1-8 doi:10.1063/1.4720576
Herrero CP, Ramirez R (2010) Diffusion of hydrogen in graphite: a molecular dynamics simulation. J Phys D: Appl Phys 43:255402-1-7 doi:10.1088/0022-3727/43/25/255402
Ferro Y, Marinelli F, Allouche A (2002) Density functional theory investigation of H adsorption and H2 recombination on the basal plane and in the bulk of graphite: connection between slab and cluster model. J Chem Phys 116:8124–8131
Dino WA, Miura Y, Nakanishi H, Kasai H, Sugimoto T (2003) Stable hydrogen configurations between graphite layers. J Phys Soc Jpn 72:1867–1870
Lamari FD, Levesque D (2011) Hydrogen adsorption on functionalized graphene. Carbon 49:5196–5200
Tozzini V, Pellegrini V (2011) Reversible hydrogen storage by controlled buckling of graphene layers. J Phys Chem C 115:25523–25528
Sofo JO, Chaudhari AS, Barber GD (2007) Graphane: a two-dimensional hydrocarbon. Phys Rev B 75:153401-1-4 doi:10.1103/PhysRevB.75.153401
Haile JM (1992) Molecular dynamics simulation: elementary methods. Wiley, New York
Simon JM, Haas OE, Kjelstrup S (2010) Adsorption and desorption of H2 on graphite by molecular dynamics simulations. J Phys Chem C 114:10212–10220
Henwood D, David Carey J (2007) Ab initio investigation of molecular hydrogen physisorption on graphene and carbon nanotubes. Phys Rev B 75:245413-1-10 doi:10.1103/PhysRevB.75.245413
Dimitrakakis GK, Tylianakis E, Froudakis GE (2008) Pillared graphene: a new 3-D network nanostructure for enhanced hydrogen storage. Nano Lett 8:3166–3170
Reina A, Jia X, Ho J, Nezich D, Son H, Bulovic V, Dresselhaus MS, Kong J (2009) Large area, few-layer graphene films on arbitrary substrates by chemical vapor deposition. Nano Lett 9(1):30–35
Zhao H, Min K, Aluru NR (2009) Size and chirality dependent elastic properties of graphene nanoribbons under uniaxial tension. Nano Lett 9:3012–3015
Poirier E, Chahine R, Benard P, Cossement D, Lafi L, Melancon E, Bose TK, Desilets S (2004) Storage of hydrogen on single-walled carbon nanotubes and other carbon structures. Appl Phys A 78:961–967
Wang L, Stuckert NR, Yang RT (2011) Unique hydrogen adsorption properties of graphene. AICHE J 57:2902–2908
Tibbetts GG, Meisner GP, Olk CH (2001) Hydrogen storage capacity of carbon nanotubes, filaments, and vapor-grown fibers. Carbon 39:2291–2301
Zhu HW, Ci LJ, Chen A, Mao ZQ, Xu CL, Xiao X, Wei BQ, Liang J, Wu DH (2000) Hydrogen uptake in multi-walled carbon nanotubes at room temperature. In: Mao ZQ, Veziroglu TN (eds) Hydrogen energy progress XIII, Proceedings of the 13th world hydrogen energy conference. International association for hydrogen energy, Beijing, pp 560–564
Wu HB, Chen P, Lin J, Tan KL (2000) Hydrogen uptake by carbon nanotubes. Int J Hydrogen Energy 25:261–265
Browning DJ, Gerrard ML, Laakeman JB, Mellor IM, Mortimer RJ, Turpin MC (2000) Investigation of the hydrogen storage capacities of carbon nanofibres prepared from an ethylene precursor. In: Mao ZQ, Veziroglu TN (eds) Hydrogen energy progress XIII, Proceedings of the 13th world hydrogen energy conference. International association for hydrogen energy, Beijing, pp 554–559
Gupta BK, Awasthi K, Srivastava ON (2000) New Carbon Variants: Graphitic nanofibres and nanotubules as hydrogen storage materials. In: Mao ZQ, Veziroglu TN (eds) Hydrogen energy progress XIII, Proceedings of the 13th world hydrogen energy conference. International association for hydrogen energy, Beijing, pp 487–492
Darkrim F, Levesque D (2000) High adsorptive property of opened carbon nanotubes at 77 K. J Phys Chem B 104:6773–6776
Phillips AB, Shivaram BS (2008) High capacity hydrogen absorption in transition metal-ethylene complexes observed via nanogravimetry. Phys Rev Lett 100:105505-1-4 doi:10.1103/PhysRevLett.100.105505
Kalamse V, Wadnerkar N, Chaudhari A (2013) Multi-functionalized naphthalene complexes for hydrogen storage. Energy 49:469–474
Phillips AB, Shivaram BS, Myneni GR (2012) Hydrogen absorption at room temperature in nanoscale titanium benzene complexes. Int J Hydrogen Energy 37:1546–1550
Kalamse V, Wadnerkar N, Deshmukh A, Chaudhari A (2012) Interaction of molecular hydrogen with Ni doped ethylene and acetylene complex. Int J Hydrogen Energy 37:5114–5121
Kalamse V, Wadnerkar N, Chaudhari A (2010) Hydrogen storage in C2H4V and C2H4V+ organometallic compounds. J Phys Chem C 114:4704–4709
Kalamse V, Wadnerkar N, Deshmukh A, Chaudhari A (2012) C2H2M (M = Ti, Li) complex: a possible hydrogen storage material. Int J Hydrogen Energy 37:3727–3732
Acknowledgments
This work was supported by the National Science Council of Taiwan under grants NSC 100-2628-E-151-003-MY3 and NSC 100-2221-E-151-018-MY3.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wu, CD., Fang, TH., Lo, JY. et al. Molecular dynamics simulations of hydrogen storage capacity of few-layer graphene. J Mol Model 19, 3813–3819 (2013). https://doi.org/10.1007/s00894-013-1918-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-013-1918-5