
Abstract
In this work, the third derivative of the energy with respect to the number of electrons, the so-called hyper-hardness, is investigated to assess whether this quantity has a chemical meaning. To achieve this goal a new working expression for hyper-hardness is developed and analyzed. It transpired from this analysis that hyper-hardness, just like hardness, can measure the reactivity or the stability of electron systems. Interestingly, positive values of hyper-hardness point to quite stable species such as noble gases and molecules. On the other hand, radicals almost always display large negative values of hyper-hardness.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the main goals of theoretical chemistry is to understand and predict the chemical behavior of chemical compounds. Along the years, different concepts have been developed to provide the necessary tools to rationalize the outcome of a chemical reaction. Among these tools, one of the most important is electronegativity [1]. The concept of electronegativity has strongly evolved from its early wording by Berzelius [2] in the nineteenth century to its absolute definition in the 1960s by Izckowski and Margrave [3]. Eventually, a quantum justification of the concept of electronegativity has been put forward by Parr and co-workers [4] and constitutes the cornerstone of the conceptual density functional theory [5, 6]. Electronegativity is nowadays defined as the opposite of the first derivative of the energy with respect to the number of electrons.
Following the definition of absolute electronegativity, the next step was to focus on the second derivative of the energy with respect to the number of electrons. Thus, this index has been identified in a seminal contribution by Parr and Pearson [7] to the chemical hardness. This identification has open wide a new field of investigation. For instance, the hard and soft acids and bases principle [8, 9] has provided a new paradigm [10–12]. Another example is the proposal for a maximum hardness principle [13]. Actually, the importance of hardness, through its reciprocal, has been recognized earlier by the work of Huheey [14, 15] and later by Politzer [16, 17] and was called at that time charge capacity. Charge capacity has been successfully used to understand the chemical behavior of fluorine and classical organic groups such as methyl, ethyl, amino, nitro and others [18].
It is quite interesting to notice that the next derivative, called the hyper-hardness, has been paid very little attention so far. Presumably because it is generally admitted that third derivatives of the energy, at the notable exception of the dual descriptor [19, 20], are supposedly [21], not that relevant for understanding chemical reactivity. Actually, the title of the present article refers to a paper by Geerlings and De Proft that reviewed this topic [22]. Thus, following the pioneering spirit of Huheey and Politzer, it has been decided to investigate the third derivative of the energy with respect to the number of electrons to check whether hyper-hardness possesses a chemical meaning. Aiming to this goal, in the section Another working equation for hyper-hardness, a new working equation for this descriptor has been developed from the hardness functional proposed by Chattaraj, Parr and Cedillo [23]. Then, in the section Chemical meaning of hyper-hardness, several arguments have been put forward to propose a chemical meaning for hyper-hardness. The next section investigates the relation between hyper-hardness and the Maximum Hardness Principle. Eventually, the values of hyper-hardness, borrowed from Parr and Fuentealba [24], for most of the chemical elements are discussed within proposed chemical meaning. Finally, hyper-hardness values calculated for some molecules and radical are examined in sections Hyper-hardness values for molecules and Hyper-hardness values for radicals. The paper ends with some concluding remarks.
Another working equation for hyper-hardness
There are three preconceptions attached to hyper-hardness. The first one is that hyper-hardness values are quite small almost zero. The second, that those values are very likely negative. Finally, to our best knowledge no chemical meaning has been found yet for hyper-hardness. It is also possible that hyper-hardness has none. In this part those preconceptions are revisited. To reach this goal, a working equation for hyper-hardness is developed.
The definition of the hyper-hardness in the canonical ensemble is [21, 25, 26]:
Two notations exist for hyper-hardness. The original one, γ , is now regularly replaced by the more modern η(2) that emphasizes its definition as the derivative of the hardness with respect to the number of electrons. Hence, hyper-hardness can also be called second order hardness. To get a working equation for the hyper-hardness, several ways have already been explored [27]. To our best knowledge the use of the N-derivative of the hardness functional has never been tested. Hardness can be calculated by minimizing the hardness functional [23] defined by Chattaraj, Cedillo and Parr as:
In equation (2), η(r,r′) stands for the hardness kernel while g(r) is any trial function normalized to 1. The minimization of this functional yields an important result: g(r)=f(r). The function f(r) is the actual Fukui Functions [28] (FF). As a result hardness can be calculated through:
Hyper-hardness can therefore be computed by taking derivatives of Eq. (3) with respect to the number of electrons at constant external potential:
Which reads:
In Eq. (5), Δf(r) represents the dual descriptor [29] (DD). Generally, the hardness kernel is decomposed into two different parts; the Coulomb contribution which is independent of the external potential and is supposed to be predominant; and the exchange-correlation contribution supposedly negligible but likely to be dependant of the external potential through its dependence to the density [30]. As a consequence, the first term of Eq. 5 is probably zero or pretty small and therefore Eq. (5) can be reduced to:
This equation is easily computable, but does not give any hints about the chemical significance of the hyper-hardness. For a better understanding of the chemical meaning of the previous equation, one uses the finite difference approximation for both the FF and the DD to express the hyper-hardness in term of frontier orbitals. The FF and the DD are written in terms of electrophilic and nucleophilic Fukui functions through:
By substitution of (7) and (8) into (6) one gets:
As hardness kernel is a symmetrical function under the exchange of its coordinates [31], Eq. (9) is reduced to:
And finally by identifying the electrophilic and nucleophilic Fukui functions to the densities of the LUMO and the HOMO respectively:
In the next part, we will analyze the above expressions to propose a physical meaning for the hyper-hardness.
Chemical meaning of hyper-hardness
One way to look at the physical significance of Eq. (10) is to identify either term to the left and right N-derivative of the chemical potential. Indeed, if one introduces either the electrophilic or nucleophilic Fukui function in Eq. 3 one gets:
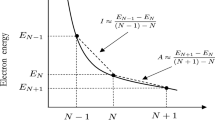
In Eqs. 12 and 13, I and A respectively represent the ionization potential and the electro-affinity. The working equation for the hyper-hardness is therefore the difference between the left and the right N-derivative of the hardness. By identification with Eq. 10 one gets:
Stating the obvious, hyper-hardness gives a comparison between the variation of the ionization potential and the electron-affinity when the number of electron of the system changes.
The question of the chemical relevance of γ has not been tackled yet. To achieve this goal, one analyses the different terms of Eq. (14).
-
The first term, \( \iint {\eta (r,r'){f^{+}}(r){f^{+}}(r')drdr'} \) describes how the electron-affinity is changing when the number of electrons evolves. This can also be seen as the part of the energy the system has to absorb to accommodate an additional electron in the LUMO.
-
The second term, \( -\iint {\eta (r,r'){f^{-}}(r){f^{-}}(r')drdr'} \) describes how the ionization potential is modified under the modification of the number of electrons. It can be seen as the part of the relief energy the system experience when it loses one electron from the HOMO.
So restricted to the frontier orbitals, the first term is therefore an indicator of the ability of the system to acquire an electron. The lower the first term, the better electrophile the system. On the contrary, the second term is an indicator of how easily the system loses an electron. The higher the second term, the better nucleophile the system. One can easily see the tendency of both terms to act in opposite directions so that the value of the hyper-hardness will be quite difficult to interpret. For instance a large negative value of γ can come from either a low value of the first term or a large value of the second term. Which means the system is either a good electrophile or a good nucleophile. The only possible interpretation is therefore that large negative values of γ indicate a system prone to react either as electrophile or nucleophile. On the contrary, low values of γ can arise when the system is neither a good electrophile (large values of the first term) nor a good nucleophile (low values of the second term). In this context, low values or positive values point to very stable systems. Consequently, one can postulate that γ is, similar to the hardness, an indicator of reactivity or stability of a system. Systems that exhibit low or positive value of γ are stable while systems with large negative values of γ are reactive. Unfortunately, and contrary to the dual descriptor, hyper-hardness, cannot discriminate nucleophilic versus electrophilic reagents. Obviously the same arguments can be applied to the hypersoftness. However, the comparison of the individual value of each component of γ would give interesting information about the ability of the system to act either as electrophile or nucleophile.
Hyper-hardness and the maximum hardness principle
Except for the electronegativity equalization principle, the other so-called DFT-based principles do not have safe and sound grounds. For instance, the maximum hardness principle (MHP), even regularly used for interpreting chemical reactions, has only been proved under the drastic conditions of chemical potential and external potential constancy. The maximum hardness principle states that molecules tend to be as hard as possible. This principle is only rigorously applicable to static systems. However, it has regularly been employed to chemical reactions with fair success, even though its application can sometimes be tricky. For instance, in the problem at hand, as the values of γ are allegedly negative, the hardness would only increase when systems loose their electrons (dη = γdN). Thus most of the systems would be qualified as nucleophile. The thesis defended herein is more complex. Indeed, during standard chemical reactions, a reagent acts either as electrophile or as nucleophile and neither as both an electrophile and a nucleophile. In this context, the hyper-hardness responds by either the first or the second of its component. During an attack by a nucleophile, the main response would be \( \iint {\eta (r,r'){f^{+}}(r){f^{+}}(r')drdr'} \) while the response to an attack by an electrophile would be \( -\iint {\eta (r,r'){f^{-}}(r){f^{-}}(r')drdr'} \).
In this context, the variation of the hardness would be positive regardless of the kind of chemical attack:
-
* \( d\eta \approx d{N^{+}}\iint {\eta (r,r'){f^{+}}(r){f^{+}}(r')drdr'} > 0 \) since both dN+ and \( \iint {\eta (r,r'){f^{+}}(r){f^{+}}(r')drdr'} \) are positive values
-
* \( d\eta \approx -d{N^{-}}\iint {\eta (r,r'){f^{-}}(r){f^{-}}(r')drdr'} > 0 \) since dN- is negative and \( \iint {\eta (r,r'){f^{-}}(r){f^{-}}(r')drdr'} \) is positive.
Another way to look at the relation between the MHP and the hyper-hardness is to use Eq. 6 into the variation of the chemical hardness:
which can be rewritten as:
As is now well established, the sign of the dual descriptor in a specific location characterizes its chemical behavior. Positions exhibiting negative values of the dual descriptor (Δf(r) < 0) are nucleophile and tend to loose electron density (δρ(r) < 0). On the other hand, positions displaying positive values of the dual descriptor (Δf(r) > 0) are electrophile and generally acquire electron density (δρ(r) > 0). In either case the chemical hardness increases which is in agreement with the MHP.
Interpretation of hyper-hardness values along the periodic table
Very few attempts have been made to calculate the values of the hyper-hardness [32]. The main reason is that there were no underlying theory to interpret the results. As a consequence the available values of hyper-hardness are quite rare in the literature. In this section, we borrowed the results from Fuentealba and Parr [24] who have calculated hyper-hardness for almost all the elements of the Periodic table. They have computed hyper-hardness from the electron-affinity (A), and the first and second ionization potential (I1 and I2), assuming that the energy varies with the number of electrons according to the relation:
In which the coefficients a, b and c are related to A, I1 and I2 through:
The chemical potential, hardness and hyper-hardness are given by:
Factors 2 and 3 have been used in Eqs. 20b and 20c to provide the values corresponding to the modern definitions of hardness and hyper-hardness. The values of all the descriptors are given in Table 1. It must be notice that few discrepancies appear in the values. The striking example is the electronegativity (opposite of chemical potential) of nitrogen that is clearly overestimated. Presumably, this is the consequence of two different causes. First of all, the experimental electron affinities of some atoms, such as nitrogen, are still unknown [33], generally just because the measurement involves the formation of an unstable anion. In Fuentealba and Parr’s publications the corresponding electron affinities have been tuned to zero. Obviously the use of negative electron affinities could address the problem. The second likely reason is the arbitrary mathematical expression taken for the dependency of the energy with respect to the number of electrons. Other functions could also have been used. However, one assumes that these flaws barely affect the third derivative of interest, namely the hyper-hardness.
It is interesting to notice first that most of the values provided by Fuentealba and Parr are negative. However some positive values are also encountered for very specific systems mostly if not all correlated to negative electroaffinities set to zero. For instance, except for helium, all the hyper-hardness values for noble gases are positive. The values are −0.73, 0.33, 0.77 and 0.61 for helium, neon, argon and xenon respectively. It is well known that noble gases are chemically stable. So, the interpretation that positive values of the hyper-hardness are associated with stable systems seems to hold. Other elements also display positive values of hyper-hardness. Those values are encountered for elements with specific electronic configuration. For instance all alkaline earth metals (Be, Mg, Ca, Sr, Ba) exhibit positive value of hyper-hardness. Those elements have their ns shell saturated. One observes the same tendency for zinc, cadmium and mercury, in which the nd shell is also saturated. On the other hand, the theory developed in this paper can not account for the fact that positive values are also encountered for scandium, yttrium and phosphorus, even though phosphorus has its np shell half saturated, but the same would apply for nitrogen which displays a negative value of the hyper-hardness.
For all the other families, the hyper-hardness values are negative. The observed tendency, expect for alkali metals, is that the values decrease when one goes down the classification. The theory defended in this paper is that high values of the hyper-hardness characterize a marked behavior of the chemical species to act as either electrophile or nucleophile. Therefore one can interpret that going down the classification, the elements are less and less prone to act as electrophile or nucleophile. Up in the periodic table the chemical behavior of atoms can be clearly defined as electrophilic or nucleophilic, while down in the periodic table, the element can change their chemical behavior according to the chemical partner they are reacting with.
Hyper-hardness values for molecules
Hyper-hardness has been calculated for several molecules using Parr and Fuentealba methodology. As hyper-hardness is defined as the third derivative of the energy with respect to the number of electrons at constant external potential, the computational method is described below. All the chemical systems have been fully optimized and characterized by frequency calculations. Then single point calculations have been performed at frozen geometry with N+1, N-1 and N-2 electrons, in which N stands for the number of electrons of the investigated system. All the computations have been performed with G09 package taking advantage of the recently introduced M062X functional which is particularly well fitted for molecules and radical calculations. The basis set used, 6-311+G(d,p), ensures that accurate energies are reached. All those values are gathered in Table 2. The molecules have been sorted out in three different kinds. The first group gathers methane and some derivatives. Benzene and benzene derivatives constitutes the second group, and finally carbonyl compounds belong to a third group.
Within each groups of molecules, some tendencies can be observed. Thus, it can be seen that for methane family, methane itself displays the highest positive value of hyper-hardness. It is well known that this compound does not exhibit any tendency to act either as an electrophile or a nucleophile. On the other hand, according to hyper-hardness values, one observes that the reactivity increases from the chloro-derivative to the iodo-derivatives. This is in agreement with what is experimentally known about akyl-halides, which reactivity follows the increasing weakness of the carbon-halide bond. It is interesting to point out that ethanamine displays the lower values of hyper-hardness and is a well-known reactive nucleophile. The values of hyper-hardness for methane family exhibit a clear pattern in which stable species have high positive values while reactive species show low positive values.
The second bunch of molecules gathers benzene derivative. Benzene and its derivative are prone to undergo electrophilic aromatic substitution (EAS). The substituent borne by the benzene has a great influence on the EAS rate. Electron withdrawing groups deactivate the aromatic ring while electron donating groups activate it. The molecules under investigation have been chosen to span the whole space of reactivity from the deactivated benzaldehyde to the very reactive styrene. Again the values of hyper-hardness translate their relative stability or reactivity. As can be seen in Table 2, taking benzene as a reference, the deactivated benzaldehyde displays a higher hyper-hardness than benzene. On the other hand, toluene, phenol and to a larger extent styrene, exhibit a lower hyper-hardness than benzene. The latter molecules are substituted by electron donating groups that increase their reactivity toward EAS. Again, hyper-hardness reflects the stability or the reactivity of a molecule.
The third group of molecule is constituted by carbonyl compounds that generally undergo nucleophilic attack on the carbon atom of the carbonyl group. The reactivity of this carbon is easily controlled by the substituent connected to the carbonyl group. For instance, one knows that saponification is several time quicker when one uses an anhydrous acid instead of an acid. The experimental reactivity sequence observed for those compounds is:
Hyper-hardness values have been calculated for methanal, acetone, acetic acid, ethanamide and urea. As can be seen in Table 2, the values of hyper-hardness continuously increase from methanal to urea, translating this way the increasing stability within this sequence. Thus, the hyper-hardness values are also able to characterize the reactivity or the stability of carbonyl compounds.
As a conclusion for this part, it can be stated that the hyper-hardness values for molecules are able to describe their relative reactivity and stability even though they are not able to ascribe whether the molecules are electrophile or nucleophile. In the next section the same investigation is undertaken for radical species.
Hyper-hardness values for radicals
Hyper-hardness values for radicals have been calculated the same way as for molecules (see Table 3). It is worth noticing that most of the hyper-hardness values for the radicals are negative whereas those of molecules are positive. This is certainly due to the fact that molecules are generally more stable than radicals. However, there is one exception. Indeed, the hyper-hardness for tertiobutylcarbonylmethyl is large and positive (0.134). Interestingly, experimental investigations indicate that this radical along with cyanomethyl is regarded as borderline since its nucleophilicity or electrophilicity is uncertain [34]. Some authors have found these radicals electrophile [35] while other authors consider them as nucleophile [36]. All other radicals display negative values of hyper-hardness. They are sorted out by decreasing hyper-hardness which means by increasing reactivity. Roughly, this classification is in agreement with the one proposed by Vleeschouwer et al. [37] based upon the electrophilicity index values. Highly reactive radicals, such as hydroxypropyl and hydroxyl, exhibit a large negative value of hyper-hardness. Mildly reactive radicals, such as tosyl or benzyl, display quite low values of hyper-hardness. There are of course some disagreements with the classification given by Vleeschouwer et al. especially, for weak or moderate reactivity. However, the general tendencies are recovered. The main drawback is that hyper-hardness only indicates whether the radical is reactive but cannot ascribe its electrophilicity or nucleophilicity.
Conclusions
In this paper, the chemical meaning of hyper-hardness has been investigated. To achieve this goal a new and computable working equation for hyper-hardness has been developed starting from the hardness functional. From this equation and using finite difference approximation, the working equation has been interpreted as the difference between the self interaction energy of one electron described by the LUMO and one electron described by the HOMO. Then it has been shown how hyper-hardness is related to the maximum hardnes principle. Besides, some arguments have been put forward to interpret the values of hyper-hardness. Systems that display positive values of hyper-hardness seems to exhibit a high stability, while large negative values are generally found for system that are chemically reactive with a strong tendency to act either as electrophile or nucleophile. This interpretation has been confronted to hyper-hardness values along the periodic table with some success. Still some positive values affected to yttrium, phosphorus and scandium can not be rationalized within this theory. Hyper-hardness values can also account for the reactivity difference within molecule families, as well as for radical species. Indeed, it has been found that radicals generally display negative values translating their great reactivity, while molecules exhibit moderate positive values. The hyper-hardness values are generally in agreement with the experimentally observed reactivity.
References
Pauling L (1932) J Am Chem Soc 54:3570–3582
Jensen MQ (1996) J Chem Educ 73:11
Iczkowski RP, Margrave JL (1961) J Am Chem Soc 83:3547–3551
Parr RG, Donelly RA, Levy M, Palke WE (1978) J Chem Phys 68:3801–3807
Geerlings P, De Proft F, Langenaeker W (2003) Chem Rev 103:1793–1873
Chermette H (1999) J Comput Chem 20:129–154
Parr RG, Pearson RG (1983) J Am Chem Soc 105:7512–7516
Pearson RG (1968) J Chem Educ 45:981–984
Pearson RG (1997) Chemical Hardness. Wiley-VCH, Weinheim
Chattaraj PK, Lee H, Parr RG (1991) J Am Chem Soc 113:1855–1856
Ayers PW, Parr RG, Pearson RG (2006) J Chem Phys 124:194107
Chattaraj PK, Ayers PW, Melin J (2007) Phys Chem Chem Phys 9:3853–3856
Pearson RG (1968) J Chem Educ 45:581–586
Huheey JE (1965) J Phys Chem 69:3284–3291
Huheey JE, Watts JC (1971) Inorg Chem 10:1553–1554
Politzer PJ (1987) Chem Phys 86:1072–1073
Politzer P, Huheey JE, Murray JS, Grodzicki MJ (1992) Mol Struct 259:99–120
Huheey JE (1971) J Org Chem 36:204–205
Morell C, Grand A, Toro-Labbé A (2005) J Phys Chem A 109:205–212
Morell C, Grand A, Toro-Labbé A (2006) Chem Phys Lett 425:342–346
Fuentealba P, Cedillo M (1999) J Chem Phys 110:9807–9811
Geerlings P, De Proft F (2008) Phys Chem Chem Phys 10:3028–3042
Chattaraj PK, Cedillo A, Parr RG (1995) J Chem Phys 103:7645–7646
Parr RG, Fuentealba P (1991) J Chem Phys 94:5559–5564
Senet P (1996) J Chem Phys 105:6471–6489
Senet P (1997) J Chem Phys 107:2516–2524
Dunlap BI (2008) J Chem Phys 129:244109
Parr RG, Yang W (1984) J Am Chem Soc 106:4049–4050
Morell C, Ayers PW, Grand A, Chermette H (2011) Phys Chem Chem Phys 13:9601–9608
De Proft F, Geerlings P, Liu S, Parr RG (1999) Polish J Chem 72:1737–1746
Berkowitz M, Parr RG (1988) J Chem Phys 88:2554–2557
Gutierrez-Oliva S, Jaque P, Toro-Labbé A (2000) J Phys Chem A 104:8955–8964
See for instance CRC Handbook of Chemistry and Physics, CRC Press
Giese B, He J, Mehl W (1988) Chem Ber 121:2063–2066
Heberger K, Lopata A (1998) J Org Chem 63:8646–8653
Wu JQ, Beranek I, Fisher H (1995) Helv Chim Acta 78:194–214
De Vleeschouwer F, Van Speybroeck V, Waroquier M, Geerlings P, De Proft F (2007) Org Lett 9:2721–2724
Acknowledgments
All the authors thank the joint program between “Evaluation-orientation de la Coopération Scientifique” and “Comisión Nacional de Investigación Científica y Tecnológica” (ECOS-CONICYT (through action project n°C11E03. C.M. and A.G. thank “le Commissariat à l’Energie Atomtique” (CEA-Grenoble) and “Grand Equipement National de Calcul Intensif” (GENCI) through projects gen6836 and gen6834 for computational support. A.T.L. acknowledges support from “Fondo Nacional de Desarrollo Científico y Tecnológico” (FONDECYT) through project n° 1090460. H.C gratefully acknowledges the “Grand Equipement National de Calcul Intensif./Centre Informatique National de l’Enseignemenet Supérieur” (GENCI/CINES) for HPC resources/computer time (Project cpt2130). Finally the authors specially thank the reviewer for pointing out the discrepancy in electronegativity values.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Morell, C., Grand, A., Toro-Labbé, A. et al. Is hyper-hardness more chemically relevant than expected?. J Mol Model 19, 2893–2900 (2013). https://doi.org/10.1007/s00894-013-1778-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-013-1778-z