
Abstract
We have investigated the adsorption properties of acetone on zigzag single-walled BNNTs using density functional theory (DFT) calculations. The results obtained show that acetone is strongly bound to the outer surface of a (5,0) BNNT on the top site directly above the boron atom, with a binding energy of −96.16 kJ mol−1 and a B–O binding distance of 1.654 Å. Our first-principles calculations also predict that the ability of zigzag BNNTs to adsorb acetone is significantly stronger than the corresponding ability of zigzag CNTs. A comparative investigation of BNNTs with different diameters indicated that the ability of the side walls of the tubes to adsorb acetone decreases significantly for nanotubes with larger diameters. Furthermore, the stability of the most stable acetone/BNNT complex was tested using ab initio molecular dynamics simulation at room temperature.

First-principles calculations predict that acetone is strongly bound to the outer surfaces of BNNTs with a binding energy of −107.14 kJ mol−1. Comparison with the corresponding adsorption on CNTs reveals that the ability of BNNTs to adsorb acetone is about threefold that of CNTs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ever since the discovery of carbon nanotubes by Iijima [1], these nanometer-scaled one-dimensional structures have been the focus of extensive investigations by the materials science community. There is particular interest in using carbon nantubes (CNTs) as sensor elements. It has been shown that the presence of gases such as NO2, NH3, and O2 can change the electrical conductivities of carbon nanotubes by several orders of magnitude, even at low gas concentrations [2, 3], which substantially exceeds the performance of comparable solid-state sensors.
In recent years, there has been increasingly interest in research and development activities related to the detection of acetone [4–15], mainly because acetone is a polar organic solvent and a major chemical commodity [4]. On the other hand, acetone is reported to be the dominant non-methane organic atmospheric pollutant [5]. Furthermore, it forms in the body during ketogenesis, leading to ketoacidosis, which is an issue for people with diabetes [6, 7].
Recently, experiments were carried out to investigate the adsorption of acetone on carbon nanotubes [8–15]. Chakrapani [14] and Shih [15] studied the interaction of acetone with SWCNTs and multiwalled carbon nanotubes (MWCNTs). They reported maximum desorption energies of about 100 kJ mol−1 and 68 kJ mol−1 for SWCNTs [14] and MWCNTs [15], respectively. Robinson et al. [8] estimated an adsorption energy of about 41 kJ mol−1 for the adsorption of acetone on CNTs. Furthermore, they showed that defects play an important role in the binding of acetone to CNTs [8]. Theoretically, the adsorption of acetone on the side-wall defects of a CNT involves a binding energy of about 34 kJ mol−1 [8].
In spite of numerous experimental and theoretical works that have presented promising results, the search is still on for a CNT material that efficiently adsorbs acetone. The solution to this problem will undoubtedly be found during the development of acetone sensors based on new nanostructured materials that offer the advantages of greater sensitivity, shorter response time, and lower cost [8–13]. By analyzing works already published in this field [16–19], we found that boron nitride nanotubes (BNNTs), which have been successfully synthesized [20–22], show enhanced adsorption capacities due to their unique properties, such as hardness, high thermal stability, and chemical inertness [16, 17, 23–28]. BNNTs are isoelectronic with CNTs, but have much wider band gaps that depend weakly on the diameter, chirality, and number of the walls of the tube [23, 24]. These properties prompted us to investigate the properties of BNNTs in acetone adsorption.
In this work, we report the results of our theoretical studies of acetone adsorption on a zigzag (5,0)/(10,0) single-walled BN nanotube (SWBNNT), and discuss its potential applications, considering that it was found to possess a faster response and a substantially higher sensitivity than those of existing solid-state sensors. The computational approach used to calculate the binding energies and the method used to construct acetone/BNNT complexes are described in detail in the next section.
Computational methods
Structural optimization and total energy calculations were performed using the ab initio DFT code SIESTA [29, 30]. We used the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) for the exchange-correlation potential [31]. The core electrons were represented by improved Troullier–Martins pseudopotentials, and a numerical atomic orbital basis with polarization was used for the valance electrons. All total energy calculations were done with a double-ζ plus polarization (DZP) basis set.
To determine the adsorption energies, the basis set superposition error (BSSE) was calculated for the optimized SWBNNT–acetone systems. This correction was performed using the counterpoise method with “ghost” atoms according to the following equation:
where E(BNNT–Acet) is the total energy of the BNNT with an adsorbed acetone molecule, while E(BNNT –Acet ghost) and E(BNNTghost–Acet) are the total energies of the nanotube and acetone alone, respectively, including the acetone and nanotube basis functions without any atomic potential.
We employed a supercell approach in all of our calculations. The unit cell of a (5,0) SWBNNT consisting of a ring of ten nitrogen and ten boron atoms with a diameter of about 3.618 Å was repeated three times along the tube axis. In the direction perpendicular to the tube axis, a distance of at least 16 Å was maintained between repeated units to avoid interactions between adjacent BNNTs. The charge density was calculated in a regular real space grid with a cutoff energy of 140 Ry. We used a 1 × 1 × 7 Monkhorst–Pack grid for k-point sampling of the Brillouin zone. The positions of all atoms were relaxed using the conjugate gradient (CG) algorithm until all of the force components were smaller than 0.02 eV Å−1.
Results and discussion
We first investigated the adsorption of acetone on the outer surface of the (5,0) SWBNNT. As shown in Fig. 1, five nonequivalent sites were considered as initial positions for the acetone molecule that is bound to the BNNT wall via its carbonyl (C=O) active site. The initial positions were above a hollow site (Hol) at the center of a hexagon, above a B–N bond at a bridge (B, N–Ax) or zigzag (B, N–Zig) site, and above a “top site,” either a boron atom (B–Top) or a nitrogen atom (N–Top). The interaction of the acetone molecule with the exterior wall of the BNNT was studied by performing single point energy (SPE) calculations for the five orientations of the axis of the molecule described above. The system included 30 B atoms, 30 N atoms, and one acetone molecule.

Models for five different adsorption states associated with the adsorption of an acetone molecule on the side wall of a (5,0) BNNT. In the adsorption states, the acetone molecule is initially located above a a boron atom (B–Top), b a nitrogen atom (N–Top), c a B–N bond at a bridge site (B,N–Ax), c a B–N bond at a zigzag (B,N–Zig) site, and e a hollow site in the center of a hexagon (Hol). Atoms are color-coded: gray carbon, white hydrogen, blue nitrogen, red oxygen, light brown boron
Optimized (5,0) BNNT and acetone molecules were used to investigate the adsorption of acetone. To find a good approximation of the stable adsorption configuration, the structures of the tube and the acetone molecule were fixed while the distance between the tube and the molecule was varied, allowing the system energy to be obtained as a function of the separation. Figure 2 shows the calculated adsorption energy (binding energy) of the system as a function of the distance between the acetone molecule and the surface of the wall for various orientations.

Binding energy of an acetone molecule as a function of the distance between the exterior surface of the (5,0) BNNT and the acetone atom closest to that surface (O) for the five molecular orientations shown in Fig. 1
The results obtained showed that the adsorption energy was slightly dependent on the orientation and location of the acetone molecule, and that the interaction rapidly became repulsive as the molecule approached the nanotube side wall. Full structural optimization was carried out for the most stable configuration of each of the systems considered. The calculated adsorption energies indicated that the acetone molecule is preferentially adsorbed at the top site directly above the boron atom (configuration B–Top). The calculated binding energy for the most energetically favorable configuration and the equilibrium distance between the (5,0) BNNT and the acetone atom closest to the BNNT were about −0.93 eV (−96.16 kJ mol−1) and 1.654 Å, respectively. The results of our first-principles calculation also showed that the bond length of C=O in acetone was 1.237 Å, which is larger than that in an isolated molecule (1.208 Å). The small separation of the adsorbed acetone from the (5,0) BNNT surface, the high negative adsorption energy of −96.16 kJ mol−1, and the increase in the C=O bond length in the adsorbed acetone all indicate a strong interaction (chemisorption) of acetone with the (5,0) BNNT [32–45]. Further information on the bond length of the isolated acetone molecule and that of the acetone adsorbed onto the nanotube is shown in Fig. 3. The binding energies obtained after full structural optimization of other considered states were found to range between −0.70 and −0.80 eV.

The optimized geometric structures of the most stable states of a isolated acetone and b the acetone/(5,0) BNNT system. c Snapshot of the DFT-based MD simulation of the most stable acetone/(5,0) BNNT structure at 300 K and 1.5 ps
We now present the results of our ab initio molecular dynamics (MD) simulation of the most stable acetone/BNNT complex, which was done in order to test whether the system studied here is stable. We performed MD simulation at room temperature for 1,500 time steps, with each step taking 1 × 10−15 s. We observed that the system is quite stable at room temperature, with an average B–O equilibrium distance of 1.488 Å, which is smaller (more stable) than the optimized system. A snapshot of the DFT-based MD simulation of the most stable acetone/(5,0) BNNT complex at 300 K and 1.5 ps is presented in Fig. 3b.
For comparison, the adsorption of an acetone molecule on a single-wall (5,0) CNT was also studied using a similar approach. Four possible configurations were selected where the O atom is pointing toward the surface of the tube. These positions were: above a hollow site (Hol) in the center of a hexagon, above a C–C bond at a bridge (C,C–Ax) or a zigzag (C,C–Zig) site, and above a “top site,” directly above a carbon atom (C–Top). The optimized (5,0) CNT and acetone molecules were used to investigate the molecular adsorption. Figure 4 shows the calculated adsorption energy of the system as a function of the distance between the acetone molecule and the surface of the CNT wall these four orientations. The most stable adsorption site for the acetone molecule was the center of a hexagon of carbon atoms. To further investigate the adsorption of the acetone on the CNT, we carried out full structural optimization of the most stable configurations. The results showed that the bond length of C=O (in acetone) was 1.209 Å, which is the basically the same as that for an isolated acetone molecule (1.207 Å). The calculated binding energy E ads and equilibrium C=O distance (C atom from the CNT and O atom from the acetone) were about 0.14 eV and 2.589 Å, respectively. The positive binding energy and the large molecule–nanotube distance reveal that there is only a weak interaction between the CNT and acetone during the adsorption. Figure 5a presents the optimized geometric structure of the most stable acetone/CNT complex.

Binding energy of an acetone molecule to a (5,0) CNT as a function of the distance from the exterior surface of the (5,0) CNT to the closest acetone atom (O) to the surface for the four considered configurations

Two orientations (a side view and b front view) of the optimized geometric structure of the most stable state of the acetone/(5,0) CNT complex. c Snapshot of the DFT-based MD simulation of the most stable acetone/CNT structure at 300 K and 1.5 ps
To evaluate the stability of the most stable acetone/CNT complex, we carried out a similar ab initio MD simulation at room temperature. The results obtained indicated that acetone was still physisorbed on the tube wall, but the geometry of the adsorbed acetone was different: its molecular axis became parallel with the nanotube axis, as depicted in Fig. 5b. Furthermore, as the figure shows, the acetone prefers to interact with the nanotube surface via both of its carbonyl and alkyl groups at room temperature.
A comparison of the calculated binding energies and binding distances for the adsorption of acetone on the BNNT and CNT showed that the binding energy of acetone on the BNNT was much higher than that on the CNT, while the binding distance of the acetone on the BNNT was shorter than that on the CNT. The shorter binding distance and higher binding energy indicate that BNNTs are more able to adsorb acetone than carbon nanotubes are.
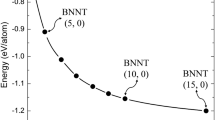
We then evaluated the effect of BNNT curvature (diameter) on acetone adsorption. A (10,0) BNNT was selected for this purpose, and similar calculation procedures were performed for an acetone molecule attached to the various active sites on the tube. After full structural optimization of all five of the configurations considered, the calculated adsorption energies were found to be positive for all of the systems considered, indicating that the acetone binds only very weakly to this large-diameter BNNT. Figure 6 shows the optimized geometric structure of the most stable state (adsorption energy of about 0.05 eV) of the acetone/(10,0) BNNT complex. Upon comparing the results obtained for the (5,0) and (10,0) BNNTs, we can conclude that the binding energy for the adsorption of acetone on a BNNT decreases with increasing BNNT diameter (i.e., the resulting state becomes thermodynamically less stable).

Optimized geometric structures of the most stable states of a the acetone/(10, 0) BNNT complex and b the acetone/(5,0) BNNT complex where the BNNT is 24.169 Å in length
We also examined the influence of nanotube length on the acetone adsorption. A (5,0) BNNT with double the length (24.169 Å) previously employed was considered for the sake of comparison. After full structural optimization of the system, the tube length was found to exert no significant effect on the adsorption ability of the tube (the adsorption energy was the same as that seen for the shorter BNNT). Figure 6b presents the optimized geometric structure of the most stable long-tube (5,0) BNNT/acetone system.
To gain a deeper understanding of the interaction between acetone and the BNNT (CNT), we also analyzed the density of states (DOS) for the combined acetone/BNNT (acetone/CNT) system and compared it with the corresponding DOSs for the individual parts, i.e., the DOS of the BNNT (CNT) and the DOS of the acetone molecule. Figure 7 shows the total electronic DOS for the most stable state of the acetone/(5,0) CNT (Fig. 7a) and acetone/(5,0) BNNT complexes (Fig. 7b). It can be seen from the figures that the DOS of the acetone/CNT system is almost exactly the superposition of the DOSs of the acetone and CNT. This finding again indicates that the acetone and CNT interact rather weakly, and that no significant hybridization of the respective orbitals of the two entities takes place; the small interaction is obtained quantitatively in terms of binding energies.

Comparison of the density of states (DOS) for a an isolated acetone molecule, an isolated CNT, and the acetone/CNT system at equilibrium geometry, and b an isolated acetone molecule, an isolated BNNT, and the acetone/BNNT system at equilibrium geometry. All energies were reduced with respect to the Fermi energies of the considered systems
However, for the acetone/(5,0) BNNT system, the results obtained show that the DOS below the Fermi level is affected by the adsorption of acetone at the BNNT surface (Fig. 7b). The results of the calculations also reveal that the DOS of the BNNT shifts up by about 0.20 eV in comparison with that of a bare BNNT when the acetone is adsorbed. This rather substantial shift can be explained by the reduction in effective Coulomb potential caused by the charge transfer. On the other hand, the difference in the Fermi levels of the BNNT (E F = −4.12 eV) and acetone/BNNT (E F = −3.74 eV) clearly shows that charge transfer occurs between the BNNT and acetone during the adsorption process. Hence, we performed Mulliken charge analyses to evaluate the amount of electron transfer between the BNNT and acetone molecules. Charge analysis showed that 0.51 e of charge were transferred from the acetone molecule to the BNNT during acetone/BNNT complex formation, whereas about 0.08 e were transferred from the acetone molecule to the CNT during acetone/CNT complex formation. The study of electronic structure and Mulliken analysis emphasize that BNNTs and acetone interact more strongly than CNTs and acetone.
To gain further insight into the acetone adsorption of BNNTs and CNTs, total electron density maps of the combined systems were calculated. Figure 8 presents calculated isosurface maps for the acetone/(5,0) or (10,0) BNNT and acetone/(5,0) CNT systems. It is clear that for the acetone/(10,0) BNNT and acetone/(5,0) CNT complexes (Fig. 8a, b), the physically adsorbed acetone is far from the cage and therefore has almost no effect on the electronic charge distribution across the B/N and C atoms in the tube, which means that no significant charge transfer occurs between the two interacting components of the system. However, for the acetone/(5,0) BNNT complex (Fig. 8c), strong hybridization between the O and B atoms is observed, which results in significant charge transfer in the system.

Isosurface maps of the total electron density for a acetone/(10,0) BNNT, b acetone/(5,0) BNNT, and c acetone/(5,0) CNT complexes (0.07 was used as the isovalue for the total electron density)
Conclusions
We investigated the interaction of an acetone molecule with a zigzag single-walled (5,0) CNT and (5,0) BNNT by using a first-principles density functional theory (DFT) method. Four or five possible configurations were selected for the approach of the acetone molecule to the carbon/boron nitride nanotube substrate. The calculated results showed that the acetone molecule binds more strongly to the outer surface of the (5,0) BNNT at the top site directly above a boron atom, with the binding energy of about −96.16 kJ mol−1. A study of the electronic structure suggested mixing of the electron states of the BNNT and acetone molecules during the adsorption process. This adsorption resulted in strong B–O bonding and charge transfer from the acetone molecule to the BNNT. The results obtained for the acetone/CNT system showed that there was only a very weak interaction between the acetone and nanotube in this case.
We also investigated the effect of tube diameter on acetone adsorption onto BNNTs, and the results obtained showed that acetone molecules bind more strongly to the exterior surfaces of small-diameter BNNTs.
Electronic structural analyses and Mulliken analyses highlighted that the acetone molecule and (5,0) BNNT interact strongly (chemisorption). However, acetone interacts only very weakly with the CNT and (10,0) BNNT (i.e., physisorption)—no significant hybridization between their respective orbitals takes place.
An ab initio DFT–MD simulation carried out at room temperature showed that the most stable acetone/(5,0) BNNT complex is quite stable and that is possible to adsorb acetone using zigzag (5,0) BNNTs. Based on our first-principles simulation results, we predict that zigzag (5,0) BNNTs are better binding materials for acetone than CNTs are. These theoretical results should be confirmed experimentally.
References
Iijima S (1991) Nature (London) 354:56
Kong J, Franklin NR, Zhou C, Chapline MG, Peng S, Cho K, Dai H (2000) Science 287:622
Collins PG, Bradley K, Ishigami M, Zettl A (2000) Science 87:1801
Kirk RE, Othmer DF, Kroschwitz JI (2007) Kirk–Othmer concise encyclopedia of chemical technology, 5th edn. Wiley, New York
Singh HB, Ohara D, Herlth D, Sachse W, Blake DR, Bradshaw JD, Kanakidou M, Crutzen PJJ (1994) Geophys Res (Atmos) 99(D1):1805
VanItallie TB, Nufert TH (2003) Nutr Rev 61(10):327
Federici MO, Benedetti MM (2006) Diabetes Res Clin Pract 74:S77
Robinson JA, Snow ES, Badescu SC, Reinecke TL, Perkins FK (2006) Nano Lett 6(8):1747
Lu Y, Partridge C, Meyyappan M, Li JJ (2006) Electroanal Chem 593(1–2):105
Snow ES, Perkins FK (2005) Nano Lett 5(12):2414
Parikh K, Cattanach K, Rao R, Suh D-S, Wu A, Manohar SK (2006) Sens Actuators B Chem B113(1):55
Guirado-Lopez RA, Sanchez M, Rincon ME (2007) J Phys Chem C 111(1):57
Kazachkin D, Nishimura Y, Irle S, Morokuma K, Vidic RD, Borguet E (2008) Langmuir 24:7848
Chakrapani N, Zhang YM, Nayak SK, Moore JA, Carroll DL, Choi YY, Ajayan PM (2003) J Phys Chem B 107(35):9308
Shih Y-H, Li M-S (2008) J Hazardous Mater 154(1–3):21–28
Wu X, An W, Zeng XC (2006) J Am Chem Soc 128(36):12001
An W, Wu X, Yang JL, Zeng XC (2007) J Phys Chem C 111(38):14105
Chen X, Wu P, Rousseas M, Okawa D, Gartner Z, Zettl A, Bertozzi CR (2009) J Am Chem Soc 131(3):890
Blase X, Rubio A, Louie SG, Cohen ML (1994) Europhys Lett 28:335
Chopra NG, Luyken RJ, Cherrey K, Crespi VH, Cohen ML, Louie SG, Zettl A (1995) Science 269:966
Hernandez E, Goze C, Bernier P, Rubio A (1998) Phys Rev Lett 80:4502
Chang CW, Han WQ, Zettl A (2005) Appl Phys Lett 86:173102
Rubio A, Corkill JL, Cohen ML (1994) Phys ReV B 49:5081
Bengu E, Marks LD (2001) Phys Rev Lett 86:2385
Xiang HJ, Yang JL, Hou JG, Zhu QS (2003) Phys Rev B 68:035427
Loiseau A, Willaime F, Demoncy N, Hug G, Pascard H (1996) Phys Rev Lett 76:4737
Wanga R, Zhu R, Zhang D (2008) Chem Phys Lett 467:131
Wang R, Zhang D (2008) Aus J Chem 61(12):941
Ordejón P, Artecho E, Soler JM (1996) Phys Rev B 53:10441
Soler JM, Artecho E, Gale JD, Garcýa A, Junqera J, Ordejón P, Sanchez-Portal D (2002) J Phys Condens Matter 14:2745
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865
Lu AJ, Pan BC (2005) Phys Rev B 71:165416
Ganji MD (2008) Phys Lett A 372:3277
Ganji MD (2008) Nanotechnology 19:025709
Gowtham S, Scheicher RH, Pandey R, Karna SP, Ahuj R (2008) Nanotechnology 19:125701
Pupysheva OV, Farajian AA, Yakobson BI (2008) Nano Lett 8:3
Ganji MD (2009) Diamond Related Mater 18:662
Ganji MD, Tajbakhsh M, Laffafchi M (2010) Sol State Sci 12:1547
Ganji MD, Mirnejad A, Najafi AA (2010) Sci Tech Adv Mater 11:045001
Ganji MD (2009) Phys E 41:1433
Ganji MD, Ahmadian N, Goodarzi M, Khorrami HA (2011) J Comp Theor Nanosci 8(8):1392–1399
Ganji MD, Mohseni M, Goli O (2009) J Mol Structure: Theochem 913:54
Ganji MD (2010) Fullerenes Nanotubes Carbon Nanostruct 18:24
Ganji MD, Rezvani M, Shokry M, Mirnejad A (2011) Fullerenes Nanotubes Carbon Nanostruct 19:421
Ahmadian N, Darvish Ganji M, Laffafchi M (2012) Mater Chem Phys 135:569
Acknowledgments
The authors gratefully acknowledge the support of this work provided by the Azad University of Ghaemshahr.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ganji, M.D., Rezvani, M. Boron nitride nanotube based nanosensor for acetone adsorption: a DFT simulation. J Mol Model 19, 1259–1265 (2013). https://doi.org/10.1007/s00894-012-1668-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-012-1668-9