
Abstract
The aim of the present research was to study the anticancer effects of Aspergillus niger (A.niger) RNase. We found that RNase (A.niger RNase) significantly and dose dependently inhibited invasiveness of breast cancer cell line MDA MB 231 by 55 % (P < 0.01) at 1 μM concentration. At a concentration of 2 μM, the anti invasive effect of the enzyme increased to 90 % (P < 0.002). Keeping the aim to determine molecular level interactions (molecular simulations and protein docking) of human actin with A.niger RNase we extended our work in-vitro to in-silico studies. To gain better relaxation and accurate arrangement of atoms, refinement was done on the human actin and A.niger RNase by energy minimization (EM) and molecular dynamics (MD) simulations using 43A2 force field of Gromacs96 implemented in the Gromacs 4.0.5 package, finally the interaction energies were calculated by protein-protein docking using the HEX. These in vitro and in-silico structural studies prove the effective inhibition of actin activity by A.niger RNase in neoplastic cells and thereby provide new insights for the development of novel anti cancer drugs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Non-communicable diseases, like cancer, heart disease and cerebrovascular disease, remain the leading causes of death, together accounting for 58 % of all deaths. Cancer is a leading cause of death worldwide, it accounted for 7.9 million deaths (around 13 % of all deaths) each year and is projected to continue rising, with an estimated 17 million deaths in 2030. About 72 % of all cancer deaths in 2007 occurred in low- and middle-income countries. Computational methods play an increasingly important role in drug designing activities. Among them, comparative protein modeling, molecular dynamics simulation and protein docking play a major role in this challenge.
The presence of actin rich pseudopods have been described as a prerequisite for cancer-cell functions. When compared to other chemotherapeutic drugs, onconases are reported to be less cytotoxic and very weakly immunogenic in clinical trials [1]. They have been demonstrated to exert both cytotoxic and cytostatic activities on mammalian cells [2] and also suppress proliferation of cancer cells in vitro [1, 3] and in vivo by increasing the cytotoxicity of several anticancer agents [4–6]. Currently phase III clinical trials are undergoing for onconases as an anticancer drug for the treatment of malignant mesothelioma [7]. When incubated with human breast cancer cell line for, e.g., MDA MB 231 cells in culture, Asperigillus niger Ribonuclease (A.niger RNase) showed both cytostatic and anti- invasive activities. It was observed that A.niger RNase binds to cell surfaces leading to inhibition of cell motility and invasiveness through matrigel coated filters. Actibind has been reported to possess antiangiogenic and anti-carcinogenic characteristics in vitro and reduced tumor size in athymic mouse xenograft models [8].
RNase displays a variety of biological functions such as degradation of RNA, control of gene expression, cell growth and differentiation, cell protection from pathogens and apoptosis etc. Besides, RNases works as potential anti-tumor drugs due to their cytotoxicity and uniquely influences several functions in the tumor cells. Microbial RNases are considered alternatives to chemotherapeutic drugs because of their ability to inhibit animal tumors and viruses; thus, recently proposed as potential anticancer agents [9–11]. Similarly, the antiviral, antifungal activities of RNases have been reported [12–15]. RNases have also been used commercially in the production of nucleotides for clinical and seasoning purposes [16]. It has been observed earlier that the various RNases from different sources such as onconases (Rana pipiens), bovine seminal RNase (Bovine seminal fluid), RNase T1 (Aspergillus oryzae), α-sarcin (Aspergillus giganteus), RNase P (Cultured human cells), ACTBIND (Aspergillus niger B1 (CMI CC 324626)) and RNase T2 (Cultured human cells) have been used for the treatment of cancer.
Molecular dynamic (MD) simulations are the interface between theory and experiment at the crossroad of pharmacy, biology, chemistry, mathematics, physics and computer science. In our previous work [17], the 3D models of human actin and A.niger RNase were designed with homology modeling. The present study was designed to gain better relaxation and accurate arrangement of atoms. Refinement was done on the built models of human actin and A.niger of RNase by energy minimization (EM) and MD simulations using 43a1 force field of Gromacs96 implemented in the Gromacs 4.0.5 package. Finally the interaction energies of human actin and A.niger RNase were calculated by protein-protein docking using the HEX.
Materials and methods
In vitro studies
MDA-MB-231 human breast carcinoma cell line was obtained from the American Type Culture Collection (Rockville, MD). The cell line was maintained in tissue culture medium (Leibovitz-15 medium supplemented with 10 % heat-inactivated fetal bovine serum, 2 mM L-glutamine, lx nonessential amino acids, 50 IU/mL penicillin, and 50 IU/mL streptomycin) at 37 °C and 5 % of CO2.
Effect on tumor cell invasion through matrigel
MDA MB 231 cells cultured in six-well plates were treated with different concentrations (1 μM and 2 μM) of A.niger RNase [18] for 2 days, released from the plates to form a single cell suspension by a brief exposure to trypsin-EDTA (Invitrogen). The cells were then washed and resuspended in serum-free medium at 2.5 × 103 cells per well. Conditioned medium of MDA MB 231 breast cancer cells was added to the lower chamber as a chemo attractant. Cell invasion was tested using Biocoat Matrigel invasion chambers after incubating for 20 h at 37 °C and 5 % of CO2. Cells that had not penetrated the filter were wiped out with cotton swabs and cells that had migrated to the lower surface of the filter were stained with giemsa, examined by bright field microscopy and photographed. The data were expressed as average number of migrated cells from 5 fields in each of the three experiments done.
Colony-formation assay
Human breast cancer cells (MDA MB 231) were grown in six- well plates (104 cells per well). Each well contained 2 mL of Leibovitz-15 medium supplemented with 10 % fetal bovine serum (FBS), 1 % glutamine, and 1 % antibiotic-antimycotic solution in the presence or absence of 1 μM, 2 μM and 3 μM ACTIBIND. The breast cancer cells were also exposed to the same concentration of enzymatically inactivated (EI)-ACTIBIND, which was preheated for 45 min in an autoclave (120 °C, 120 kPa) until no RNase activity was detected. The cells were incubated at 37 °C. After 48 h, 5000 cells/plate were seeded in 35 × 10 mm Petri plates and were allowed to grow for 7 days. The cells were then fixed in 4 % formaldehyde and stained with methylene blue. The experiment was done in triplicates and the number of colonies was counted by two individuals separately who were blinded to the experiment.
Cytologic observations
Human breast cancer cells (MDA MB 231) were cultured in the presence and absence of 1 μM and 2 μM RNase. The breast cancer cells were also exposed to the same concentration of enzymatically inactivated (EI)-RNase. Cells were fixed, stained with Mayer hematoxylin and eosin (H & E) and observed under inverted microscope. The experiments done were in triplicates.
In-silico studies
All the calculations of MD simulation and analysis were performed on the ENSEMBL of system configurations on a workstation AMD Opteron Duo-core 2.0 GHz and 4 GB RAM. MD simulations were analyzed with Gromacs 4.0.5; protein-protein docking calculations were performed with HEX. In our previous work molecular modeling tasks were performed with Modeller9v5; for modeling of the human actin and A.niger RNase the following crystallographic structures were used as templates, 3D3Z, 2BTF, respectively [17]. The modeling procedure begins with the alignment of the sequence to be modeled (target) with relatively known three-dimensional structures (templates). This alignment is usually the input to the program and the output is a three-dimensional model (3D) for the target sequence containing all main-chain and side-chain non-hydrogen atoms.
Molecular dynamics and simulation studies
In order to validate the sequence-structure alignment, to remove bad contacts derived from the homology modeling and to achieve a good starting structure, the model was subjected to exhaustive molecular dynamics simulation upto 8 ns with Gromacs 4.0.5 software using Gromacs 96.1 (43 A2) force field [19]. The initial structure was placed in a solvent box (7.552 nm × 6.339 nm × 8.751 nm) with a total volume of 418.93 nm3. The box was filled with water (11,397 molecules). Each structure was placed in the center of a truncated cubic box filled with extended simple point charge (SPC/E) water molecules. The initial simulation cell dimensions were 79.54 Å, 83.76 Å and 51.79 Å for the system, and had the protein solvated by a layer of water molecules of at least 10 Å in all directions in the system. In the MD protocol, all hydrogen atoms, ions, and water molecules were first subjected to 200 steps of energy minimization by the steepest descent algorithm [20] to remove close van der Waals contacts. The system was then submitted to a short MD simulation with position restraints for a period of 1 ps and afterward it was subjected to a full MD without restraints. The temperature of the system was then increased from 50 to 310 K in five steps (50–100 K, 100–150 K, 150–200 K, 200–250 K, and 250–310 K) and the velocities at each step was reassigned according to the Maxwell-Boltzmann distribution at that temperature and equilibrated for 2 ps. Energy minimization and MD were carried out under periodic boundary conditions. The simulation was computed in the isobaric-isothermal (NPT) ENSEMBL at 310 K with the Berendsen temperature coupling and a constant pressure of 1 atm with isotropic molecules-based scaling [21, 22] was maintained. The LINCS algorithm [23], with a 10 − 5 Å tolerance, was applied to fix all bonds containing a hydrogen atom, allowing the use of a time step of 2 fs in the integration of the equations of motion. No extra restraints were applied after the equilibration phase. The convergence of simulation was analyzed in terms of the potential energy, root-mean-square deviation (RMSD) from the initial model structure and root-mean- square fluctuation (RMSF). The analysis was calculated relative to the Cα back bone structures, and all coordinate frames from the trajectories were first superimposed on the initial conformation to remove any effect of overall translation and rotation.
Protein-protein docking
Docking studies were conducted to evaluate the predictive ability of the A. niger RNase homology model and its relevance for use in the structure-based drug design studies. In order to perform protein-protein docking between the models of A.niger RNase and human actin, generated model (after the molecular dynamics and simulation) PDB’s were submitted separately to online server HEX. The parameters used for the docking process were correlation type – shape only, calculation device- GPU, number of solutions-100, FFT mode –3D fast lite, grid dimension–0.6, receptor range–180, ligand range–180, twist range–360, distance range–40. HEX works on FFT correlation using spherical polar coordinates and Gaussian density representation of protein shape. The computational part of the server consists of a 32-node cluster running the CentOS 5.2 operating system and using the OAR batch scheduling system (http://oar.imag.fr/). Each node consists of two quad-cores Intel Xeon 2.5 GHz CPUs, and eight of the nodes are equipped with two Nvidia Tesla C1060 GPUs. Hence, a total of 256 CPU cores and 16 GPUs are currently available on HEX server [24]. The drug and its analogues were docked with the receptor using the above parameters. All docking poses were analyzed and visualized with Pymol and VMD [25]. There have been many efforts to predict protein-protein interaction binding sites based on the analysis of the protein surface properties [26–33] analyzed the surface patches using six parameters: solvation potential, residue interface propensity, hydrophobicity, planarity, protrusion and solvation accessible surface area. The six parameters were then combined into a global score that gave the probability of a surface patch forming protein-protein interaction.
Results and discussion
Effect of A. niger RNase in vitro
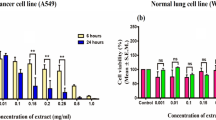
Effect on tumor cell invasion through Matrigel: RNase significantly and dose dependently inhibited invasiveness of breast cancer cell line MDA MB 231 by 55 % (P < 0.01) at 1 μM concentration. At a concentration of 2 μM, the anti invasive effect of the enzyme was found to have increased to 90 % (P < 0.002) (Fig. 1).

Effect on tumor cell invasion through Matrigel. A.niger RNase was found to bind actin. It also bound to cancer cell surfaces, leading to disruption of the internal actin network and inhibiting cell motility and invasiveness through Matrigel-coated filters
Colony-formation assay
We assessed the effect of RNase on the colony forming ability of human breast cancer cell line MDA MB.231. A. niger RNase significantly inhibited colony formation of the breast cancer cell line tested. The anticlonogenic effect was significant only in RNase pre-treated cells and was much less effective in cells that were not previously exposed to RNase. In MDA MB 231 breast cancer cells treated with different concentrations (1 μM, 2 μM and 3 μM) of A. niger RNase, the number of colonies were inhibited by 37 %, 80 %, and 97 % respectively compared with controls (Fig. 2). Inactive A. niger RNase enzyme showed a similar effect in breast cancer cells, i.e., 24 %, 75 %, and 92 % of inhibition respectively compared with controls (Table 1). The peripheral cells of the control colonies displayed numerous cytoplasmic extensions, whereas in the A. niger RNase treated colonies, the peripheral cell extensions were arrested (Fig. 3). These results suggest that A. niger RNase treatment effects the morphology of the cells that may inhibit the cancer cell motility and thereby the metastatic capacity of the cancer cell.

Effect of A.niger RNase on colony-formation. The effect of native A.niger RNase on clonogenicity in MDA MB 231 breast cancer cell lines and enzyme inactivated. The number of colonies in native A.niger RNase (red hatched square), EI- A.niger RNase (blue hatched square) than in controls (solid square)

The effect of A. niger RNase on the morphology and motility of MDA MB 231 breast cancer cells. In the control cells, fully developed cell extensions were observed in control. In A.niger RNase (1 μM and 2 μM concentrated) treated cells, the cell extensions were inhibited
Cumulatively, the evidence leads us to propose that cell-surface actin may be a target for A.niger RNase in cancer cells. Therefore, we envision that A.niger RNase may compete with angiogenin for cell-surface actin and, in this manner, blocks the formation of the actin-angiogenin complex required for cancer cell organization and angiogenesis in developing neoplastic tissue.
Analysis of model
The overall stereochemical quality of the final models for each enzyme of the human actin and A.niger RNase were assessed by the program PROCHECK, Whatif, ProSA, ERRAT, and Verify 3D, and the objective function supplied by the program MODELLER [17].
Analysis of the MD simulation
MD simulations were performed to determine the stability of the predicted 3D structure of the human actin and A.niger RNase. Analysis of 5 ns dynamics showed that the human actin and A.niger RNase structures were stable, after a rapid increase during the first 2500 ps, and 1500 ps respectively. The protein backbone RMSD average and standard deviation over the last 3 ns of the human actin and A.niger RNase trajectory was 0.55 ± 0.02 Å and 0.55 Å. The plot of the radius of gyration (Rg) versus time of human actin and A.niger RNase are presented in Fig. 4a,b. It is observed that Rg decreased to its minimal stable value of approximately 0.7 nm after 5 ns of simulations. These data reveal that the protein has attained stability protein and has attained stability during dynamic simulations. Berendsen thermostat have limitations of maintaining velocity rescaling approaches to ensure that the average kinetic energy of the system corresponding to the expected value at the desired temperature (Fig. 4c,d). The potential energy curve of human actin showed a smooth decrease until 500 ps, after which it was found fluctuating around a constant value of −2.72e + 05 kJ mol −1, A.niger RNase became stabilized at −1.53e + 05 kJ mol −1 (Fig. 4e,f). A plateau of RMSD for the human actin system was achieved within 2.7 ns of unrestrained simulation and for A.niger RNase it was 1.5 ns, suggesting that 5 ns unrestrained simulation was sufficient for stabilizing fully relaxed models. Figure 5a,b shows the evolution of the RMSD during the dynamics. In these MD simulations use of Berendsen thermostat have not been adversely affected by these limitations. The average residue fluctuations of the proteins were observed by calculating room mean square fluctuation (RMSF), RMSF values from MD of the trajectory which reflected the flexibility of each atom residue in a molecule Fig. 5c, d. The major backbone fluctuation of human actin was observed to occur in the loop regions of 40 to 50, Random coil, beta sheets and alpha helices of 170 to 210, 210 to 250, and 250 to 275 residues. Whereas regions with low RMSF corresponded exclusively to the rigid beta-alpha-beta fold, while coming to backbone fluctuation of A.niger RNase observed at the end of the chain, i.e., 19–22 residues. In a typical RMSF pattern, a low RMSF value indicates the well-structured regions while the high values indicate the loosely structured loop regions or domains terminal. In addition, analysis of the structure during the dynamics simulation indicates that the regions present higher RMSF values with 0.9 nm at loop 3, which strongly shows that these regions are the most flexible in the predicted structure of human actin.

MD simulation results. Radius of gyration, kinetic energy and potential energy (a) Radius of gyration of human actin, time (ps) verses Rg (nm), (b) Radius of gyration of A.niger RNase time (ps) verses Rg (nm), (c) Kinetic energy of human actin, E(KJmol−1) verses time(ps), (d) Kinetic energy of A.niger RNase E(KJmol−1) verses time, (e) Potential energy of human actin, E(KJmol−1) verses time(ps), (f) Potential energy of A.niger RNase E(KJmol−1) verses time(ps)

MD simulation results. RMSD & RMSF, (a) Time dependence of the RMSD for the backbone atoms of human actin, derived by molecular dynamics calculation using Gromacs software, (b) Time dependence of the RMSD for the backbone atoms of A.niger RNase derived by molecular dynamics calculation using Gromacs software, (c) of backbone atoms as a function of amino acids human actin, (d) RMSF of backbone atoms as a function of amino acids A.niger RNase
Protein-protein docking
In order to understand the inhibition mechanism of A.niger RNase on human actin, primary docking calculations were performed with HEX [17]. The goal of the initial stage of docking is to generate as many near-native complex structures (hits) as possible. The generated PDB file (after the molecular dynamics and simulation) was analyzed for their binding conformations. Analysis was based on Etotal or free energy of binding, lowest docked energy, and calculated RMSD values. For each approach, the number of hits, the RMSD value of the best hit (with the lowest RMSD) based on shape complementarity are listed in Table 2. The results obtained from protein–protein docking algorithms were satisfactory. The total clusters of docking conformations, with the top 30 docked molecules showed negative binding energies. Cluster 1 shows the energy and RMSD values to be −1.983 kcal mol−1 and 0.715 Å, respectively. Among all docking clusters, rank 10, i.e., solution 23 gave the best predicted binding free energy of −1.258 kcal mol−1 with RMSD value of 0.313 Å and docked energy of −9.529. The docking of A.niger RNase and human actin is shown in Fig. 6. A.niger RNase docking revealed that the amino acids Thr1, Leu2, Asp3, tyr5, Thr6, Ser9, Asp10, Ile13, Thr14, Pro15, Glu17, and Asp18 played vital role to bind the Tyr166, His170, Ala173, Thr351, Gln354, Glu364, Ser365, 368, Arg372, and Phe375 of human actin (Fig. 7). The ball and stick model of A.niger RNase is presented in Fig. 8. Our in-silico experiments demonstrate that A.niger RNase binds human actin, and also that itself inhibits its function and thus may act as a drug. Cumulatively, the evidence leads us to propose that cell-surface actin may be a target for RNase in cancer cells. Therefore, A.niger RNase may compete with angiogenin for cell-surface actin and, in this manner, blocks the formation of the actin-angiogenin complex required for cancer cell organization and angiogenesis in developing neoplastic tissue.

Protein-protein docking interaction of A.niger RNase represented in surface along with ball & stick form, and human actin represented in surface form along with cartoons. The image was generated using Pymol

This image presents the interacting amino acid residues on the RNase actin and RNase proteins. The interacting region of RNase is represented in different colors of ball & stick form and the regions of human actin are shown by cartoon in which catalytic residues in sticks form. The 3D structure of the RNase and actin complex was predicted by protein docking using HEX software. The image was generated using Pymol

A.niger RNase represented in ball & stick form, in which active site catalytic residues represented in colors, non interactive residues represented black color. The image was generated using Pymol
Based on the protein-protein docking results, molecular dynamics study of the human actin and A.niger RNase were performed by using the Gromacs program. First, we examined the stability of the human actin and A.niger RNase by a 2.5 ns, 1.5 ns respectively, MD simulation and following RMSD calculation. The obtained results show that human actin and A.niger RNase becomes equilibrated at 1 ns and afterward. Then the RMSDs of the human actin and A.niger RNase were obtained based on the MD simulation of systems to get information on positional fluctuations. The interaction energy of docking between the A.niger RNase and human actin was calculated and analyzed using the HEX server. The efficient binding of A.niger RNase and human actin revealed that the proteins could form hydrogen bond networks involving active amino acid residues. Several amino acid residues including Thr1, Leu2, Asp3, tyr5, Thr6, Ser9, Asp10, Ile13, Thr14, Pro15, Glu17, and Asp18 were identified to exclusively contributive to the binding of A.niger RNase to Tyr166, His170, Ala173, Thr351, Gln354, Glu364, Ser365, 368, Arg372, and Phe375 of human actin. The generated homology model is expected to be useful for the structure-based drug design against cancer.
Our results show that the docking scores follow a distribution from which such predictions are possible and in good agreement with the experimentally determined agonist bound A.niger RNase to human actin structures. Based on experimental data, molecular docking and dynamics study were performed to explore the inhibition mechanism of A.niger RNase toward human actin. Our results suggested that RNase could exactly bind to the active pocket of human actin to display inhibition; the binding mode may alter with the change of some substituents on certain positions. The RNase protein orientation in the active site can greatly affect the stability of the protein-protein complex. Although our results are based on a rather detailed system setup, to some extent taking advantage of prior experimental knowledge, we believe that the fundamental ideas of this study can serve as inspiration when dealing with protein docking to flexible receptors.
Conclusions
Findings from our study, signify the potentiality of A.niger RNase as an anticancer drug. The evidence leads us to propose that cell-surface actin may be a target for A. niger RNase in cancer cells. Therefore, A.niger RNase competes with angiogenin for cell-surface actin and in this manner, blocks the formation of the actin-angiogenin complex required for cancer cell organization and angiogenesis in developing neoplastic tissue.
References
Vasandani VM, Wu YN, Mikulski SM, Youle RJ, Sung C (1996) Molecular determinants in the plasma clearance and tissue distribution of ribonucleases of the ribonuclease A superfamily. Cancer Res 56:4180–4186
Darzynkiewicz Z, Carter SP, Mikulski SM, Ardelt WJ, Shogen K (1988) Cytostatic and cytotoxic effects of Pannon (P-30 Protein), a novel anticancer agent. Cell Tissue Kinet 21:169–182
Mikulski SM, Viera A, Darzynkiewicz Z, Shogen K (1992) Synergism between a novel amphibian oocyte ribonuclease and lovastatin in inducing cytostatic and cytotoxic effects in human lung and pancreatic carcinoma cell lines. Br J Cancer 66:304–310
Ryback SM, Pearson JW, Fogler WF, Volker K, Spence SE et al (1996) Enhancement of vincristine cytotoxicity in drug-resistant cells by simultaneous treatment with onconase, an antitumor ribonuclease. J Natl Cancer Inst 88:747–753
Deptala A, Halicka HD, Ardelt W, Mikulski SM, Shogen K et al (1998) Potentiation of tumor necrosis factor induced apoptosis by onconase. Int J Oncol 13:11–16
Matousek J, Soucek J, Slavik T, Tomanek M, Lee JE et al (2003) Comprehensive comparison of the cytotoxic activities of onconase and bovine seminal ribonuclease. Comput Biochem Physiol C 136:343–356
Mikulski SM, Costanzi JL, Vogelzang NJ, McCachren S, Taub RN et al (2002) Phase II trial of a single weekly intravenous dose of ranpirnase in patients with unresectable malignant mesothelioma. J Clin Oncol 20:274–281
Schwartz B, Shoseyov O, Melnikova VO, McCarty M, Leslie M et al (2007) ACTIBIND, a T2 RNase, competes with angiogenin and inhibits human melanoma growth, angiogenesis, and metastasis. Cancer Res 67:5258–5266
Zhang Y, Li W, Su Z (2001) Trends in the pharmaceutical research of ribonucleases and their therapeutic uses. J Biomed Eng 18:456–460
Makarov AA, Ilinkaya O (2003) Cytotoxic ribonucleases: molecular weapons and their targets. FEBS Lett 540:15–20
Makarov AA, Kolchinski A, Ilinskaya ON (2008) Binase and other microbial RNases as potential anticancer agents. BioEssays 30:789–790
Saxena SK, Gravell M, Wu Y (1996) Inhibition of HIV-1 production and selective degradation of viral RNA by an amphibian ribonuclease. J Biol Chem 271:20783–20788
Skvortsova MA, Bocharov AL, Yakovley GI (2002) Novel extracellular ribonuclease from Bacillus intermedius-binase II: purification and some properties of the enzyme. Biochem Mosc 67:802–806
Guan GP, Wang HX, Ng TB (2007) A novel ribonuclease with antiproliferative activity from fresh fruiting bodies of the edible mushroom Hypsizigus marmoreus. Biochim Biophys Acta 1770:1593–1597
Lam SK, Ng TB (2001) Isolation of a novel thermolabile heterodimeric ribonuclease with antifungal and antiproliferative activities from roots of the Sanchi Ginseng Panax otoginseng. Biochem Biophys Res Commun 285:419–423
Lindblom M, Morgen H (1974) Enzymatic RNA reduction in disintegrated cell Saccharomyces cerevisia. Biotechnol Bioeng 16:1123–1133
Gundampati RK, Rajasekhar C, Moni K, Anurag S, Pratyush DD et al (2011) Protein-protein docking on molecular models of Aspergillus niger RNase and human actin: novel target for anticancer therapeutics. J Mol Model 18:653–662
Gundampati RK, Sharma A, Kumar M, Debnath M (2011) Extracellular poly (A) specific ribonuclease from Aspergillus niger ATCC 26550: purification, biochemical, and spectroscopic studies. Process Biochem 46:135–141
Patricia CP, Rafael AC, Fernanda C, Walter FA (2009) Molecular modeling dynamics simulation of human cyclin-dependent kinase 3 complexed with inhibitors. Comput Biol Med 39:130–140
Arfken G (1985) The method of steepest descents.n§7.4. In: Mathematical methods for physicists, 3rd edn. Academic, Orlando, pp 428–436
Berendsen HJC, Grigera JR, Straatsma TP (1987) The missing term in effective pair potentials. J Phys Chem 91:6269–6271
Chowdhuri S, Tan ML, Ichiye TJ (2006) Dynamical properties of the soft sticky dipole–quadrupole–octupole water model: a molecular dynamics study. J Chem Phys 125:14451–14453
Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472
Macindoe G, Mavridis L, Venkatraman V, Devignes MD, Ritchie DW (2010) Hex server: an FFT-based protein docking server powered by graphics processors. Nucleic Acids Res 38:445–449. doi:10.1093/nar/gkq311
Humphrey W, Dalke A, Schulten K (1996) VMD-visual molecular dynamics. J Mol Graph 14:33–38
Lichtarge O, Bourne HR, Cohen FE (1996) An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol 257:342–358
Jones S, Thronton JM (1997) Analysis of protein-protein interaction sites using surface patches. J Mol Biol 272:121–132
Jones S, Thromton JM (1997) Prediction of protein protein interaction sites using patches analysis. J Mol Biol 272:133–143
Neuvirth H, Raz R, Schreiber G (2004) Promote: a structure based prediction program to indentify the location of protein-protein binding sites. J Mol Biol 338:181–199
Bradford J, Westhead D (2005) Improved prediction of protein-protein binding sites using a support vector machines approach. Bioinformatics 211:487–1494
Zhou H, Shan Y (2001) Prediction of protein interaction sites from sequence profile and residue neighbor list. Proteins 44:336–343
Espadaler J, Romero-Isart O, Jackson RM, Oliva B (2005) Prediction of protein-protein interactions using distant conservation of sequence patterns and structure relationships. Bioinformatics 21:3360–3368
Aytuna AS, Gursoy A, Keskin O (2005) Prediction of protein-protein interactions by combining structure and sequence conservation in protein interfaces. Bioinformatics 2:2850–2855
Acknowledgments
GRK, SS and NG wants to thanks University Grants Commission, Government of India for financial support and the School of Biochemical Engineering, Institute of Technology, BHU, Varanasi, India. KS wishes to thank Council of Scientific & Industrial Research, Government of India for providing financial assistance. SL wants to thank Indian Council of Medical Research (ICMR),New Delhi, Government of India for financial support and Tumour Biology Laboratory, National Institute of Pathology, Safdarjung Hospital Campus, New Delhi-110029 and RSC, CM gratefully acknowledges University Grant Commission (UGC), New Delhi (F.No.33-222/2007 (SR) 13-3-08) for financial support. The encouragement given by Sri Krishnadevaraya University, AP, India to our research work is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Gundampati Ravi Kumar, Rajasekhar Chikati and Santhi Latha Pandrangi contributed equally to this work.
Rights and permissions
About this article
Cite this article
Kumar, G.R., Chikati, R., Pandrangi, S.L. et al. Molecular docking and dynamics simulations of A.niger RNase from Aspergillus niger ATCC26550: for potential prevention of human cancer. J Mol Model 19, 613–621 (2013). https://doi.org/10.1007/s00894-012-1587-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-012-1587-9