
Abstract
The in vitro antifungal potency of six series of 4-arylthiosemicarbazides was evaluated. Two isoquinoline derivatives with an ortho-methoxy or ortho-methyl group at the phenyl ring were the most potent antifungal agents. Molecular modeling studies and docking of all 4-arylthiosemicarbazides into the active sites of sterol 14α-demethylase (CYP51), topoisomerase II (topo II), l-glutamine: d-fructose-6-phosphate amidotransferase (GlcN-6-P), secreted aspartic proteinase (SAP), N-myristoyltransferase (NMT), and UDP-N-acetylmuramoyl-l-alanine:d-glutamate ligase (MurD) indicated the importance of both structural and electronic factors in ligand recognition and thus for the antifungal effectiveness of 4-arylthiosemicarbazides. A possible antifungal target was identified (NMT) and isoquinoline-thiosemicarbazides showed more favorable affinity than the native ligand.

Electrostatic potential surface of isoquiniline derivative compound 6o with antifungal activity
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Invasive fungal infections (IFIs) are life-threatening opportunistic infections that are an increasingly important cause of morbidity and mortality in patients, especially those with compromised immune function and those hospitalized with serious underlying diseases [1, 2]. The majority of these infections are caused by Candida spp., with over 50 % due to Candida albicans [3–6]. Candida species are the fourth most common pathogens isolated from patients with nosocomial bloodstream infections in the United States and the sixth in Europe [7–9]. These fungi are responsible for various forms of disease, ranging from superficial infections of the mucosal surfaces or skin to systemic infections, which in most cases are life threatening [10]. The incidence of candidemia in the US and Europe varies between 1.9 and 11 per 100,000 inhabitants [11–13]. Mortality in patients with candidemia is high, ranging from 40 % to 60 %, with reported attributable mortality of 20–40 % [9]. In general, for treatment of an infection with Candida species, amphotericin B and azole drugs are used, but these agents are not considered to satisfy medical needs because of their toxicity, side effects, drug interactions, limited routes, and the emergence of drug-resistant and drug-low-susceptible strains [14–21]. Among these limitations, the major obstacle in the treatment of C. albicans infections is the spread of antifungal drug resistance, mainly in patients chronically subjected to antimycotic therapy, i.e., those treated with broad-spectrum antibiotics, immunosuppressive agents, anticancer, and anti-AIDS drugs [22, 23]. Considering all these factors, the identification of new antifungal small molecules is an important goal of current anti-infective research.
Recently, as a part of our efforts to develop new effective antibacterial agents in the class of thiosemicarbazide derivatives, a series of 4-arylthiosemicarbazides was synthesized and their biological potency evaluated [24]. In vitro antibacterial activity assays indicated that compounds with electron-withdrawing substituents in the para position are more effective. Furthermore, it was documented for the first time that thiosemicarbazide derivatives participate in at least two different mechanisms of antibacterial activity. One of these was identified as inhibition of topoisomerase IV, while the nature of the other could not be elucidated from the limited data collected. The binding mode of the synthesized compounds was explored by flexible molecular docking, which indicated the importance of H-bonding and electrostatic interactions between the thiosemicarbazide core and amino acid residues of the ATP binding site. To further explore the diverse biological activity of thiosemicarbazide derivatives, we focused our attention on the antifungal activity and structure-activity relationships (SAR) of the 4-arylthiosemicarbazides, those already described [24–27] and nine new derivatives, using yeast Candida as the experimental model. Since selective toxicity is fundamental to the development of anti-infective agents, cytotoxicity studies were also carried out. Although the antifungal potential of thiosemicarbazide derivatives is well-recognized [28–37], thus far no detailed studies have been conducted to determine the mechanism of action and the target proteins for their antifungal activity. As most of the existing fungal drugs are enzyme inhibitors, the second aim of the present studies was to identify the interactions of 4-arylthiosemicarbazides with antifungal drug target enzymes using in silico molecular docking. In these studies, six common and novel enzymes that were considered in antifungal studies reported in literature [38–41], were selected as targets, i.e., sterol 14α-demethylase (CYP51), topoisomerase II (Topo II), l-glutamine: d-fructose-6-phosphate amidotransferase (GlcN-6-P), secreted aspartic proteinase (SAP), N-myristoyltransferase (NMT), and UDP-N-acetylmuramoyl- l-alanine: d-glutamate ligase (MurD).
Materials and methods
Chemicals
All commercial reactants and solvents were purchased from either Sigma-Aldrich (St. Louis, MO) or Lancaster (Windham, NH) with the highest purity and used without further purification. Melting points were determined on a Fischer-Johns block and are uncorrected. Elemental analyses were determined by a AMZ-CHX elemental analyzer (within ± 0.4 % of theoretical values). IR spectra were recorded in KBr using a Specord IR-75 spectrophotometer. 1H NMR spectra were recorded on a Bruker Avance (300 MHz). Analytical thin layer chromatography (TLC) was performed on Merck 60F254 silica gel plates and visualized by UV irradiation (254 nm).
Procedure for synthesis of 4-arylthiosemicarbazides
A reaction mixture of appropriate heterocarboxylic hydrazide (0.01 mol) and related isothiocyanate (0.01 mol) was heated in an oil bath at 80 °C and the progress of the reaction was monitored by TLC. After 12 h, the reaction was completed and crude reaction mixture was washed with diethyl ether and crystallized from ethanol.
Physicochemical characterization of 1a-1 m, 3d, 3n, 4b, 4d, 4p, 4q, 5c, 5h, 5i, 5m, 5n, and 6b were presented previously [24–27]. Compounds 2c, 2h, 2m, 2n are commercially available.
Procedure for synthesis of s-triazole 6o-t
The thiosemicarbazide derivative 6o (0.01 mol) was dissolved in 2 % NaOH (10 mL) and refluxed for 2 h. After cooling, the solution was neutralized with 3M HCl. The solid formed was filtered, dried and crystallized from ethanol.
Yields and spectral characterization of new compounds are provided in Supplementary Material.
Antifungal assay
The primary screen was carried out by the disc-diffusion method using agar medium, according to the Clinical and Laboratory Standards Institute guidelines [42]. For compounds showing an inhibitory effect on the growth of the tested microorganisms—monitored as an appearance of growth inhibition zones (GIZs)—minimal inhibitory concentrations (MICs) were determined using the agar dilution method, according to Clinical and Laboratory Standards Institute guidelines [43]. The detailed procedure was described in a previous paper [31].
Cytotoxicity assay
Cell culture
Cell line L929 (ATTC® Catalog No. CCL-1, mouse fibroblasts; http://www.atcc.org) were routinely cultured in Iscove’s modified Dulbecco medium (IMDM, Cytogen, Princeton, NJ), supplemented with 10 % (v/v) fetal bovine serum (FBS, Sigma), plus 2 mM l-glutamine (Sigma), 100.0 U/ml penicillin (Sigma), 100.0 μg/ml streptomycin (Sigma), 5x10−5 M 2-mercaptoethanol (Sigma) and grown at 37 °C in a 10 % CO2 humidified environment.
Suspensions of the compounds 1f, 1h, 1m, 2h, 5h, 6b and 6o were freshly prepared before the cells were exposed, and diluted to appropriate concentrations; 1–1,250 μg/mL for 1f, 1h, 1m, 1–500 μg/mL for 2h, 5h, 6o, and 1–300 μg/mL for 6b with the culture medium (containing 2.5 % DMSO). Cells treated with 2.5 % DMSO-solvent served as a control in each experiment.
Cell viability assays
The effects of tested compounds on the viability of mouse fibroblasts L929 cells were evaluated using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. The MTT assay was used according to international standards: ISO 10993-5:2009 (Tests for in vitro cytotoxicity; http://www.iso.org/iso/catalogue_detail.htm?csnumber=36406 ). L929 cells were plated into 96-well plates at a density of 1.0 x 104/100 μl/well in culture medium and allowed to attach for 24 h before treatment. Afterwards, culture medium in the plates was replaced by 100 μl compounds suspension at concentration of 0–565 μg/ml and the cells were exposed for 24 h. Then 1 mg/ml MTT (50 μl/well) was added to each well and incubated at 37 °C, 10 % CO2 for 2 h. Mitochondrial dehydrogenases of viable cells reduce the yellowish water-soluble MTT to water-insoluble formazan crystals, which were solubilized with dimethyl sulfoxide (DMSO). The cell culture medium was aspirated cautiously, after which 150 μl DMSO was added to each well and mixed thoroughly. Optical density (OD) was read on the ELISA reader (Multiskan EX, Labsystems; http://www.mtxlsi.com/multiskan_EX.htm) at 550 nm. The results were expressed as percentage viability compared with the treated 2.5 % DMSO controls. All experiments were performed in triplicate.
Computational details
Physicochemical parameters, HOMO/LUMO maps, and conformational searches were calculated using HyperChem8.0.3 [44]. Extensive conformational searches were carried out using the molecular mechanics level with the OPLS [45, 46] force field. The most stable structures obtained were subsequently optimized to the closest local minimum at the semiempirical level using RM1 parametrization [47]. Convergence criteria were set to 0.1 and 0.01 kcal mol−1 Å−1 for OPLS and RM1 calculations, respectively. Electrostatic potentials were calculated for the geometries that resulted from docking using Gaussian 03 [48] and GaussView 5 [49] at the HF/6-31G level [50, 51].
Automated docking setup
Flexible ligand–receptor docking was performed using the AutodockVina program [52] using the default settings. Models of the sterol 14α-demethylase (CYP51), topoisomerase II (topo II), l-glutamine: d-fructose-6-phosphate amidotransferase (GlcN-6-P), secreted aspartic proteinase (SAP), N-myristoyltransferase (NMT), and UDP-N-acetylmuramoyl- l-alanine: d-glutamate ligase (MurD) binding sites based on the structure deposited in the Protein Data Bank [53] under the PDB ID 2CIB [54], 1Q1D [55], 1XFF [56], 1EAG [57], 1IYL [58], 1UAG [59] were employed. Default docking parameters and flexible space of 24 x 24 x 24 Å3 were validated by re-docking native ligand that docked exactly in the position present in the crystal structure. Subsequently, all 4-arylthiosemicarbazides were docked using same docking parameters.
Results and discussion
Chemistry
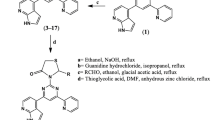
Six series (1–6) of 4-arylthiosemicarbazides were prepared in high yields according to a known procedure [24–27, 60–63] in the reaction of related heterocarboxylic acid hydrazide with aryl isothiocyanate (Scheme 1). This one-step reaction produced nine new (2i, 3h, 3m, 3q, 4k, 6k, 6n, 6o, 6p) and 30 known thiosemicarbazides derivatives (1a-1m, 2c, 2h, 2m, 2n, 3c, 3d, 3n, 4b, 4d, 4p, 4q, 5c, 5h, 5i, 5m, 5n, 6b).

Preparation of targeted 4-arylthiosemicarbazides, series 1–6. Series 1: 1a–m; series 2: 2c, 2h, 2i, 2m, 2n; series 3: 3c, 3d, 3h, 3m, 3n, 3q; series 4: 4b, 4d, 4k, 4p, 4q;series 5: 5c, 5h, 5i, 5m, 5n; series 6: 6b, 6k, 6n, 6o, 6p. For symbols used to identify studied compounds see Table 1
In vitro antifungal activity
The in vitro antifungal activity of the title 4-arylthiosemicarbazides (series 1–6) was tested in comparison with fluconazole against three strains of yeast—C. albicans ATCC 10231, C. albicans ATCC 90028 and C. parapsilosis ATCC 22019—using the agar dilution method as described in CLSI documents M7–A7 [43]. Minimal inhibitory concentrations (MICs) were defined as the lowest concentration of the compound preventing growth of the tested microorganism and are listed in Table 2. The results indicated that within series 1, compounds 1c with the para-nitro substitution, and 1f with the ortho and para positions substituted with chlorine atoms were the most potent, exhibiting moderate activity of 50 μg/mL towards C. parapsilosis. These compounds were also effective against C. albicans, however at much higher concentrations (MIC range 100–200 μg/mL). Two other derivatives; 1h with para-fluoro substitution and 1m with ortho-bromo substitution showed somewhat lower activity, whereas remaining compounds with halo substitution at the ortho and/or para position, 2,4-difluoro derivative 1i, para-bromo derivative 1k, and para-chloro derivative 1l, were inactive. When the thiadiazole scaffold was replaced with the similarly sized thiophene, pyrrole or furane (series 2, 3 and 4, respectively) antifungal potency was lost completely except at 2h. Subsequently, the antifungal potency of series 5 with an indole scaffold was studied. This search led to the identification of para-fluoro derivative 5h and 2,4-difluoro derivative 5i with MIC at 100 μg/mL towards C. parapsilosis and marginal activity towards C. albicans. Remaining derivatives with ortho-fluoro 5n, para-nitro 5c, and ortho-bromo 5m substitution were inactive. Replacement of the indole scaffold with the similarly sized isoquinoline resulted in ortho-methoxy derivative 6o with MIC at 25 μg/mL towards C. albicans and 50 μg/mL towards C. parapsilosis. An antifungal response was also observed for ortho-methyl derivative 6b, however, at somewhat higher concentration; MICs at 50 μg/mL towards all screened Candida species. Interestingly, derivatives with electron-withdrawing substitution, para-bromo derivative 6k, ortho-fluoro derivative 6n, and para-iodo derivative 6p were inactive. Finally, the biological activity of s-triazole 6o-t, a cyclic derivative of the most potent 6o, was tested. No antifungal potency was noted.
Cytotoxicity of selected compounds against L929 cells
Compounds 1f, 1h, 1m, 2h, 5h, 6b, and 6o, all of which showed in vitro antifungal activity, were further examined for their initial in vitro toxicity effects against mammalian L929 cells by MTT assay. Toxicity was defined as the highest dilution of test samples that causes 30 % or greater destruction of cells. Morphology of normal cells and cultured cells with tested compounds are given in Fig. S1 in Supplementary Material.
From a pharmacological point of view it is important for the studied compounds to exhibit high bioactivity and at the same time show no or low toxicity effects, otherwise the activity might just be due to general toxicity, which disqualifies the compound as a drug or lead molecule candidate. From a comparison of the results of toxicity collected in Fig. 1 and antifungal activity tests, it can be seen clearly that the potent anti-Candida agent 6b displays antifungal activity at non-cytotoxic concentrations in mammalian cells. Also, compounds 1f, 1h, and 2h were found to be non-toxic up to 50 and 100 μg/mL, respectively, which equals the MIC values against C. parapsilosis. Another important result from the toxicity studies is that the toxicity of the tested compounds does not seem to be correlated to their antifungal activity. For example, 1f, which was found to be non-toxic up to the same concentration as 6b (50 μg/mL) is four-time less active against Candida strains (MIC of 200 vs 50 μg/mL). Compound 1h, which was non-toxic up to 100 μg/mL, is only marginally active (MIC 400 μg/mL or higher against Candida strains). In contrast, compound 5h, which is even less antifungal than 1h was found to be non-toxic up to 10 μg/mL, while the most cytotoxic 6o (5 μg/mL) is the most potent antifungal. This indicates either that these compounds may act via different mechanisms in their antifungal activity against C. albicans and toxicity against mammalian cells or that minor structural changes afford important alterations in fungal cell permeability/transport, etc. [64]

Concentration-dependent cytotoxic activity of 1f, 1h, 1m, 2h, 5h, 6b, and 6o
Molecular modeling studies
Since the in vitro antifungal activity could not be explained reasonably in the light of theories of the influence of substituents on biological response, the difference in bioactivity of 4-arylthiosemicarbazides was therefore investigated further by a molecular modeling approach and docking studies with the hope of finding a possible explanation for the observations described above, and to provide a better understanding of the relationship between structure and antifungal activity. Initially, some structural and electronic descriptors of the title compounds were investigated (see Table S1 in the Supplementary Material). In the structure of 4-arylthiosemicarbazide, the presence of the isoquinoline ring and electron-donating methyl or methoxy groups (compounds 6b and 6o) seemed to confer higher antifungal potency than that of other substituted groups. They also are characterized by favorable E B (binding energy) values and high μ (dipole moment) and E HOMO (the highest occupied molecular orbital energy) values in contrast to all other studied compounds. Antifungal activity is probably affected by these electronic descriptors, which can be important parameters for the interaction of 4-arylthiosemicarbazides with the active sites. It is known that the energies of HOMO and lowest unoccupied molecular orbital (LUMO) orbitals serve as indices for the electron-donating and electron-acceptor abilities of the molecule, respectively. The higher the energy of the HOMO, the better its electron-donating ability. In the case of LUMO, good electron-acceptor ability is associated with low-energy molecular orbitals. In our results, antifungal potency is related to HOMO, which suggests that electron-donor ability of the molecule is important in determining activity. According to the frontier orbital maps presented in Fig. 2 for representative model compound 6o, the HOMOs of each compound were localized mainly on the sulfur atom of the thiosemicarbazide core. These observations emphasize the possible role of the sulfur atom in the charge transfer processes that occur when a ligand interacts with the biological target.

Highest occupied molecular orbital (HOMO; left) and lowest unoccupied molecular orbital (LUMO; right) contour plots for representative model compound 6o. HOMO and LUMO maps for representative model compounds series 1 and 5 are presented in our previous paper [24]
Subsequently, the localization of LUMO orbitals for each structure was analyzed. There was no quantitative correlation between antifungal activity and LUMO localization, but a trend clearly emerged; 4-arylthiosemicarbazides from series 1, 2, 5, and 6 have their LUMO situated on the heterocyclic ring, whereas LUMO density for compounds from completely inactive series 3 and 4 was distributed outside the heterocyclic moiety, mainly on the sulfur atom. This behavior might be interpreted in the sense that 4-arylthiosemicarbazides with the electron-accepting character of the heterocyclic ring at N1 position interact better with the biological target.
Finally, conformational analysis of the studied compounds was performed according to the protocol described for 4-arylthiosemicarbazides with thiadiazole (series 1) and indole (series 5) moieties [24]. As expected based on our previous studies, no structural requirements needed for antifungal activity of 4-arylthiosemicarbazides could be deduced; both isoquinoline derivatives with significant activity, 6b and 6o, as well as inactive ones 6k, 6n, and 6p shared very similar geometry. Again, comparison of the molecular shape of active thiophene derivative 2h with the most potent antifungal 6o or inactive ones 2m, 2n, 3h, 3d, 4b did not show any recognizable relationship with activity (data not shown). Thus, the present study manifested the importance of electronic properties rather than the restricted stereochemistry of the molecule for the antifungal response.
Docking studies
As noted in the Introduction, with the hope if identifying the fungal cellular targets of thiosemicarbazide class compounds, we analyzed interactions of active and inactive molecules with the active sites of common and novel enzymes that were considered in antifungal studies reported in the literature [38–41]. The following enzymes were included in the studies: sterol 14α-demethylase (CYP51), topoisomerase II (Topo II), l-glutamine: d-fructose-6-phosphate amidotransferase (GlcN-6-P), secreted aspartic proteinase (SAP), N-myristoyltransferase (NMT), and UDP-N-acetylmuramoyl- l-alanine: d-glutamate ligase (MurD). The docking simulations were performed using the program AutoDock Vina, which was applied successfully in our previous studies of the binding mode of thiosemicarbazide derivatives in the active site of bacterial topoisomerase II [24]. The best poses of the studied compounds within the active sites of target enzymes are displayed in Fig. 3 and their corresponding binding Gibbs free energies ΔGb are shown in Table 3.

Superimposition of the native ligand (rendered as tubes) and the best conformations of representative ligands 1a, 1b, 1c, 1f, 1 h, 1 m, 2c, 2 h, 3n, 3q, 4d, 4 k, 5c, 5 h, 5i, 5n, 6b, 6n, 6o, and 6p docked to the binding site of sterol 14α-demethylase (CYP51) (ID 2cib), topoisomerase II (Topo II) (ID 1q1d), secreted aspartic proteinase (SAP) (ID 1eag), and N-myristoyltransferase (NMT) (ID 1iyl)
As indicated by the positive value of the Gibbs free energy, none of the ligands bound the active sites of GlcN-6-P and MurD, while potential inhibitory activities of the studied compounds against CYP51, Topo II, SAP, and NMT were observed. Almost all best poses in the series 1–6 were located in a position very similar to that of the inhibitors on which these enzymes were crystallized. Some SAR trends were observed when the docking conformations and binding free energies of the studied compounds in the active site of NMT were analyzed. The nonpeptidic inhibitor that NMT was crystallized with contains a benzofuran core and a long aminoaliphatic chain. From among the compounds studied by us, those containing an isoquinoline ring (series 6) show more favorable binding affinity than the native ligand, and bind to the site occupied by the benzofuran ring. The remaining compounds, series 1–5, show binding Gibbs free energies lower than the native ligand, and bind to the site occupied by the aminoaliphatic chain situated at position 4 of the benzofuran ring, except for 1a, 1b, 2h, 5c, 5h, 5i, and 5m (Fig. 4). This supports the conclusion reached from experiments [58] that the part of the active site to which the benzofuran moiety is allocated is responsible mainly for binding. Another significant finding is that, with the exception of 2h and 2i, all compounds bind to the active site of NMT without H-bond interactions with amino acid residues. This is a very important result, which points to the fact that the electronic nature of a molecule may play the key role in ligand recognition.

Superimposition of the native ligand (rendered as tubes) and the best conformations of ligands from series 1–6 docked to the binding site of NMT (1iyl)
Subsequently, we focused on isoquinoline derivatives (series 6) as this series contains the two most active compounds: 6b and 6o. Based on the conformations obtained from docking studies, we generated electrostatic potential surfaces. As seen in Fig. 5, the most active compound (6o) is characterized by electrostatics around the sulfur atom that differ significantly from that in the other ligands. The is explained mainly by the opposite orientation of the proton in the neighboring NH group. Following this finding, we analyzed the geometry of this fragment of the thiosemicarbazide skeleton in detail (Table 4). The opposite location of the NH proton is characterized by the HN(N)CS dihedral angle being significantly different, and opposite in sign to that in all other compounds. The graphical representation of the different electrostatics in this part of 6o presented in Fig. 5 was quantified by the Mulliken partial atomic charge at the sulfur atom, which was also significantly different and opposite in sign than in other ligands. Two other geometric parameters, i.e., C–S bond distance and HN(C)CS dihedral angle, are also distinct in the case of 6o. It should be noted that these differences are very subtle and can be easily overlooked. The importance of electrostatics and geometry of the –NH–C(=S)–NH– fragment can be further inferred from the corresponding parameters of the methyl derivative 6b. As can be seen in Table 4, 6b is closer to 6o than to the remaining ligands. This correlates well with bioactivity, with that of 6b being lower than that of 6o and the other ligands being inactive.

Comparison of the electrostatic potential surfaces of compounds from series 6 that resulted from docking studies at the active site of NMT
Based on the above analysis, we also addressed the question of the lack of antifungal activity of s-triazole 6o-t, which is a dehydroderivative of the most active thiosemicarbazide 6o. We docked compound 6o-t to the active site of NMT. As illustrated in Fig. 6, the best pose of 6o-t shows only partial overlap with the corresponding pose of 6o, which coincides well with the position of the ligand used in crystallographic studies. Most importantly, the orientation of the N–C(=S)–N fragment of the s-triazole ring is completely different and the location of the sulfur atom within the active site is completely different, 8.7 Å away from the location of sulfur atom of 6o, which, as argued above, influences the antifungal activity of this ligand. Thus, the structural and electronic factors of the NH-NH–C(=S)–NH core seem to be important for ligand recognition and, consequently, for the antifungal effectiveness of the studied 4-arylthiosemicarbazides. Only use of specific enzymatic bioassays, will allow this hypothesis to be confirmed and the biological target of the tiosemicarbazide class of compounds to be identified unequivocally.

Superimposition of the best conformations of 6o and 6o-t docked to the binding site of NMT (1iyl)
Conclusions
The present study evaluated the in vitro antifungal activities of six series of 4-arylthiosemicarbazides. Among the compounds tested, two isoquinoline derivatives 6o and 6b were the most potent antifungals. Subsequently, molecular modeling and flexible docking studies were carried out to identify and characterize the structural and electronic properties that modulate the antifungal potency of 4-arylthiosemicarbazides. The electronic requirements for antifungal effectiveness deduced from the molecular modeling approach are (1) high HOMO and dipole moment values, (2) favorable binding energy, and (3) the presence of an electron accepting heteroaromatic ring at N1 position of thiosemicarbazide skeleton. The structural and electronic requirements for ligand recognition deduced from comparisons of ligands in the active site of NMT are (1) high-electron density around the sulfur atom, and (2) geometry of NH-NH-C(=S)-NH core. The results of the docking study of inactive s-triazoles 6o-t, a dehydroderivative of the most potent antifungal agent 6o, confirmed the validity of the pharmacophore model developed for 4-arylthiosemicarbazides, although additional experimental evidence is needed to support this hypothesis. Efforts aimed at elucidating the antifungal mechanism of action of 4-arylthiosemicarbazides using biochemical approaches are underway in our group.
References
Pfaller MA, Diekma DJ (2007) Epidemiology of invasive candidiasis: persistent public health problem. Clin Microbiol Rev 20:133–163
Negri M, Martins M, Henriques M, Svidzinski TI, Azeredo J, Oliveira R (2010) Examination of potential virulence factors of Candida tropicalis clinical isolates from hospitalized patients. Mycopathologia 169:175–182
Borg-von Zepelin M, Kunz L, Rüchel R, Reichard U, Weig M, Gross U (2007) Epidemiology and antifungal susceptibilities of Candida spp. to six antifungal agents: results from a surveillance study on fungaemia in Germany from July 2004 to August 2005. J Antimicrob Chemother 60:424–428
Mercedes Panizo M, Reviakina V, Dolande M, Selgrad S (2008) Candida spp. in vitro susceptibility profile to four antifungal agents. Resistance surveillance study in Venezuelan strains. Med Mycol 24:1–7
Richardson M, Lass-Flörl C (2008) Changing epidemiology of systemic fungal infections. Clin Microbiol Infect 14:5–24
Pappas PG, Kauffman CA, Andes D, Benjamin DK Jr, Calandra TF, Edwards JE Jr, Filler JF, Kullberg BJ, Ostrosky-Zeichner L, Reboli AC, Rex JH, Walsh TJ, Sobel JD (2009) Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 48:503–535
Pfaller MA, Diekema DJ, Jones RN, Sader HS, Fluit AC, Hollis RJ, Messer SA (2001) International surveillance of bloodstream infections due to Candida species: frequency of occurrence and in vitro susceptibilities to fluconazole, ravuconazole, and voriconazole of isolates collected from 1997 through 1999 in the SENTRY antimicrobial surveillance program. J Clin Microbiol 39:3254–3259
Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB (2004) Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39:309–317
Méan M, Marchetti O, Calandra T (2008) Bench-to-bedside review: Candida infections in the intensive care unit. Crit Care 12:204–212
Lass-Flörl C (2009) The changing face of epidemiology of invasive fungal disease in Europe. Mycoses 52:197–205
Diekema DJ, Messer SA, Brueggemann AB, Coffman SL, Doern GV, Herwaldt LA, Pfaller MA (2002) Epidemiology of candidemia: 3-year results from the emerging infections and the epidemiology of Iowa organisms study. J Clin Microbiol 40:1298–1302
Kao AS, Brandt ME, Pruitt WR, Conn LA, Perkins BA, Stephens DS, Baughman WS, Reingold AL, Rothrock GA, Pfaller MA, Pinner RW, Hajjeh RA (1999) The epidemiology of candidemia in two United States cities: results of a populationbased active surveillance. Clin Infect Dis 29:1164–1170
Tortorano AM, Kibbler C, Peman J, BernhardtH KL, Grillot R (2006) Candidaemia in Europe: epidemiology and resistance. Int J Antimicrob Agents 27:359–366
Enoch DA, Ludlam HA, Brown NMJ (2006) Invasive fungal infections: a review of epidemiology and management options. J Med Microbiol 55:809–818
Blot S, Vandewoude K (2004) Management of invasive candidiasis in critically ill patients. Drugs 64:2159–2175
Hakki M, Staab JF, Marr KF (2006) Emergence of a Candida krusei isolate with reduced susceptibility to caspofungin during therapy. Antimicrob Agents Ch 50:2522–2524
Moudgal V, Little T, Boikov D, Vazquez JA (2005) Multiechinocandin- and Multiazole-resistant Candida parapsilosis isolates serially obtained during therapy for prosthetic valve endocarditis. Antimicrob Agents Chemother 49:767–769
Arendrup MC, Fuursted K, Gahrn-Hansen B, Jensen IM, Knudsen JD, Lundgren B, Schonheyder HC, Tvede MJ (2005) Seminational surveillance of fungemia in Denmark: notably high rates of fungemia and numbers of isolates with reduced azole susceptibility. J Clin Microbiol 43:4434–4340
Hobson RPJ (2003) The global epidemiology of invasive Candida infections—is the tide turning? J Hosp Infect 55:159–168
Verweij PE, Snelders E, Kema GHJ, Mellado E, Melchers JGM (2009) Azole resistance in Aspergillus fumigatus: a side-effect of environmental fungicide use? Lancet Infect Dis 6:789–795
Groll AH, De Lucca AJ, Walsh TJ (1998) Emerging targets for the development of novel antifungal therapeutics. Trends Microbiol 6:117–124
Sheehan DJ, Hitchcock CA, Sibley CM (1999) Current and emerging azole antifungal agents. Clin Microbiol Rev 12:40–79
Schiaffella F, Macchiarulo A, Milanese L, Vecchiarelli A, Costantino G, Pietrella D, Fringuelli R (2005) Design, synthesis, and microbiological evaluation of new Candida albicans CYP51 inhibitors. J Med Chem 48:7658–7666
Siwek A, Stączek P, Stefańska J (2011) Synthesis and structure-activity relationship studies of 4-arylthiosemicarbazides as topoisomerase IV inhibitors with Gram-positive antibacterial activity. Search for molecular basis of antibacterial activity of thiosemicarbazides. Eur J Med Chem 46:5717–5726
Siwek A, Wujec M, Dobosz M, Wawrzycka-Gorczyca I (2010) Study of direction of cyclization of 1-azolil-4-aryl/alkyl-thiosemicarbazides. Heteroatom Chem 21:521–532
Siwek A, Wujec M, Stefańska J, Paneth P (2009) Antimicrobial properties of 4-aryl-(2-methyl-furan-3-yl)-Δ2-1H-1,2,4-triazole-thiones. Phosphorus Sulfur 184:3149–3159
Wawrzycka I, Siwek A, Kosikowska U, Malm A (2011) Synthesis and crystal structure of N-butyl-5-(4-methyl-1,2,3-thiadiazol-5-yl)-1,3,4-thiadiazol-2-amine and 5-isoquinolin-3-yl-N-(2-methylphenyl)-1,3,4-thiadiazol-2-amine. Z Kristallogr 226:297–302
Çapan G, Ulusoy N, Ergenç N, Kiraz M (1999) New 6-Phenylimidazo2,1-b]thiazole derivatives: synthesis and antifungal activity. Monatsh Chem 130:1399–1407
Dogan HN, Rollas S, Erdeniz H (1998) Synthesis, structure elucidation and antimicrobial activity of some 3-hydroxy-2-naphthoic acid hydrazide derivatives. Farmaco 53:462–467
Farag AM, Mayhoub AS, Barakat SE, Bayomi AH (2008) Synthesis of new N-phenylpyrazole derivatives with potent antimicrobial. Bioorg Med Chem 16:4569–4578
Siwek A, Stefańska J (2011) Antimicrobial activity and SAR study of some novel thiosemicarbazide derivatives bearing piperidine moiety. Med Chem 7:690–696
Salgın-Gökşen U, Gökhan-Kelekçi N, Göktaş Ö, Köysal Y, Kılıç E, Işık S, Aktay G, Özalp M (2007) 1-Acylthiosemicarbazides, 1,2,4-triazole-5(4H)-thiones, 1,3,4-thiadiazoles and hydrazones containing 5-methyl-2-benzoxazolinones: synthesis, analgesic-anti-inflammatory and antimicrobial activities. Bioorg Med Chem 15:5738–5751
Wujec M, Kosikowska U, Paneth P, Malm A (2007) Reaction of hydrazide of (tetrazol-5-yl)acetic acid with isothiocyanates and antimicrobial investigations of newly-obtained compouns. Heterocycles 71:2617–2626
Cunha S, Macedo FC Jr, Costa GAN, Rodrigues MT Jr, Verde RBV, de Souza Neta LC, Vencato I, Lariucci C, Sá FP (2007) Antimicrobial activity and structural study of disubstituted thiourea derivatives. Monatsh Chem 138:511–516
Kadi AA (2011) Synthesis and antimicrobial activity of some new quinazolin-4(3H)-one derivatives. J Saudi Chem Soc 15:95–100
Al-Saadi MS, Faidallah HM, Rostom SAF (2008) Synthesis and biological evaluation of some 2,4,5-trisubstituted thiazole derivatives as potential antimicrobial and anticancer agents. Arch Pharm 341:424–434
Jalilian AR, Sattari S, Bineshmarvasti M, Shafiee A, Daneshtalab M (2000) Synthesis and in vitro antifungal and cytotoxicity evaluation of thiazolo-4H-1,2,4-triazoles and 1,2,3-thiadiazolo-4H-1,2,4-triazoles. Arch Pharm 333:347–354
Ruge E, Korting HC, Borelli C (2005) Current state of three-dimensional characterisation of antifungal targets and its use for molecular modelling in drug design. Int J Antimicrob Ag 26:427–441
Sheng Ch XuH, Wang W, Cao Y, Dong G, Wang S, Che X, Ji H, Miao Z, Yao J, Zhang W (2010) Design, synthesis and antifungal activity of isosteric analogues of benzoheterocyclic N-myristoyltransferase inhibitors. Eur J Med Chem 45:3531–3540
Balladka KS, Bettadapura GK, Chenna GD, Basavapattana RB, Hanumanthappa M (2010) Synthesis, characterization, in vitro and molecular docking studies of new 2,5-dichloro thienyl substituted thiazole derivatives for antimicrobial properties. Eur J Med Chem 45:3490–3496
Khan SI, Nimrod AC, Mehrpooya M, Nitiss L, Walker LA, Clark AM (2002) Antifungal activity of eupolauridine and its action on DNA topoisomerases. Antimicrob Agents Ch 46:1785–1792
Clinical and Laboratory Standards Institute (2006) Performance standards for antimicrobial disc susceptibility tests; approved standard M2-A9. Clinical and Laboratory Standards Institute, Wayne, PA
Clinical and Laboratory Standards Institute (2006) Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard M7-A7. Clinical and Laboratory Standards Institute, Wayne, PA
HyperChem 8.0.3 (2007) HyperCube, Gainsville, FL
Jorgensen WL, Maxwell DS, Tirado-Rives J (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 118:11225–11236
Jorgensen WL, McDonald NA (1998) Development of an all-atom force field for heterocycles. Properties of liquid pyridine and diazenes. J Mol Struct (THEOCHEM) 424:145–155
Rocha GB, Freire RO, Simas AM, Stewart JJP (2006) Rm1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J Comput Chem 27:1101–1111
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian03, Revision D.01. Gaussian, Wallingford, CT
Dennington II R, Keith T, Millam J, Eppinnett K, Hovell WL, Gilliland R (2003) GaussView, Version 3.0. Semichem, Shawnee Mission, KS
Francl MM, Pietro WJ, Hehre WJ, Binkley JS, Gordon MS, Defrees DJ, Pople JA (1982) Self-consistent molecular orbital methods XXIII. A polarization-type basis set for second-row elements. J Chem Phys 77:3654–3665
Hariharan PC, Pople JA (1973) The influence of polarization functions on molecular orbital hydrogenation energies. Theor Chim Acta 28:213–222
Trott O, Olson AJ (2010) AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242; http://www.pdb.org
Podust LM, von Kries JP, Nasser Eddine A, Kim Y, Yermalitskaya LV, Kuehne R, Ouellet H, Warrier T, Alteköster M, Lee J-S, Rademann J, Oschkinat H, Kaufmann SHE, Waterman MR (2007) Small-molecule scaffolds for CYP51 inhibitors identified by high-throughput screening and defined by X-ray crystallography. Antimicrob Agents Chemother 51:3915–3923
Classen S, Olland S, Berger JM (2003) Structure of the topoisomerase II ATPase region and its mechanism of inhibition by the chemotherapeutic agent ICRF-187. Proc Natl Acad Sci USA 100:10629–10634
Isupov MN, Obmolova G, Butterworth S, Badet-Denisot MA, Badet B, Polikarpov I, Littlechild JA, Teplyakov A (1996) Substrate binding is required for assembly of the active conformation of the catalytic site in Ntn amidotransferases: evidence from the 1.8 A crystal structure of the glutaminase domain of glucosamine 6-phosphate synthase. Structure 4:801–810
Cutfield SM, Dodson EJ, Anderson BF, Moody PC, Marshall CJ, Sullivan PA, Cutfield JF (1995) The crystal structure of a major secreted aspartic proteinase from Candida albicans in complexes with two inhibitors. Structure 3:1261–1271
Sogabe S, Masubuchi M, Sakata K, Fukami TA, Morikami K, Shiratori Y, Ebiike H, Kawasaki K, Aoki Y, Shimma N, D'Arcy A, Winkler FK, Banner DW, Ohtsuka T (2002) Crystal structures of Candida albicans N-myristoyltransferase with two distinct inhibitors. Chem Biol 9:1119–1128
Bertrand JA, Auger G, Fanchon E, Martin L, Blanot D, van Heijenoort J, Dideberg O (1997) Crystal structure of UDP-N-acetylmuramoyl-l-alanine:d-glutamate ligase from Escherichia coli. EMBO J 16:3416–3425
Ezabadi IR, Camoutsis C, Zoumpoulakis P, Geronikaki A, Soković M, Glamoćilija J, Ćirić A (2008) Sulfonamide-1,2,4-triazole derivatives as antifungal and antibacterial agents: Synthesis biological evaluation, lipophilicity, and conformational studies. Bioorg Med Chem 16:1150–1161
Kuş C, Ayhan-Kılcıgil G, Özbey S, Kaynak FB, Kaya M, Çoban T, Can-Eke B (2008) Synthesis and antioxidant properties of novel N-methyl-1,3,4-thiadiazol-2-amine and 4-methyl-2H-1,2,4-triazole-3(4H)-thione derivatives of benzimidazole class. Bioorg Med Chem 16:4294–4303
Önkol T, Doğruer DS, Uzun L, Adak S, Özkan S, Şahin MFJ (2008) Synthesis and antimicrobial activity of new 1,2,4-triazole and 1,3,4-thiadiazole derivatives. Enzyme Inhib Med Chem 23:277–284
Shams HZ, Mohareb RM, Helal MH, Mahmoud AE (2007) Synthesis, structure elucidation, and biological evaluation of some fused and/or pendant thiophene, pyrazole, imidazole, thiazole, triazole, triazine, and coumarin systems based on cyanoacetic 2-(benzoylamino)thioxomethyl. hydrazide. Phosphorus Sulfur 182:237–263
Babu KS, Li XC, Jacob MR, Zhang Q, Khan SI, Ferreira D, Clark AM (2006) Synthesis, antifungal activity, and structure-activity relationships of coruscanone A analogues. J Med Chem 49:7877–7886
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Siwek, A., Stefańska, J., Dzitko, K. et al. Antifungal effect of 4-arylthiosemicarbazides against Candida species. Search for molecular basis of antifungal activity of thiosemicarbazide derivatives. J Mol Model 18, 4159–4170 (2012). https://doi.org/10.1007/s00894-012-1420-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-012-1420-5