
Abstract
Monoamine oxidase (MAO) is an enzyme of major importance in neurochemistry, because it catalyzes the inactivation pathway for the catecholamine neurotransmitters, noradrenaline, adrenaline and dopamine. In the last decade it was demonstrated that imidazoline derivatives were able to inhibit MAO activity. Furthermore, crystallographic studies identified the imidazoline-binding domain on monoamine oxidase B (MAO-B), which opens the possibility of molecular docking studies focused on this binding site. The goal of the present study is to identify new potential inhibitors for MAO-B. In addition, we are also interested in establishing a fast and reliable computation methodology to pave the way for future molecular docking simulations focused on the imidazoline-binding site of this enzyme. We used the program ‘molegro virtual docker’ (MVD) in all simulations described here. All results indicate that simplex evolution algorithm is able to succesfully simulate the protein-ligand interactions for MAO-B. In addition, a scoring function implemented in the program MVD presents high correlation coefficient with experimental activity of MAO-B inhibitors. Taken together, our results identified a new family of potential MAO-B inhibitors and mapped important residues for intermolecular interactions between this enzyme and ligands.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Monoamine oxidase (monoamine: oxygen oxidoreductase (deaminating), EC 1.3.3.4, MAO) is a flavin-dependent enzyme that catalyzes the oxidative deamination of important amine neurotransmitters, such as dopamine, noradrenaline, and serotonine. For a recent review see [1]. These important protein targets are found in the external mitochondrial membrane as two isoenzymes, MAO-A and MAO-B, that exhibit differing substrate and inhibitor specificities [2–8]. MAO-B is selective for dopamine whereas MAO-A is selective for serotonin and noradrenaline. Inhibition of MAO-A and B causes amplification of the existing amounts of monoamine neurotransmitters in the brain for the therapy of psycho-neurological disorders. Several MAO-B inhibitors are currently useful in Parkinson’s disease [5, 9, 10].
Several molecular docking studies have been performed on MAOs [11–21], none of them was focused on the imidazoline binding site on MAO-B, also known as I2 binding site [22–26]. Although experimental evidence suggested the importance of this binding site for inhibitory activity against MAO-B [27–30], that could be used for molecular docking studies. Recent X-ray crystallographic and biochemical studies [31] revealed the structural basis for an interesting potentiation of MAO-B inhibition due to the presence of a ligand in the substrate binding site and 2-(2-Benzofuranyl)-2-imidazoline (2-BFI) in the imidazoline site. Analysis of the structure of the 2-BFI complex with tranylcypromine-inhibited MAO-B showed that the presence of tranylcypromine modifies the positioning of Leu199 in the entrance of the active site, generating a closed conformation for MAO-B. This form allows tight binding of the reversible inhibitor 2-BFI with 1000 fold increase in the affinity [31]. Furthermore, this study suggested that a new generation of MAO-B inhibitors could be obtained focusing on the entrance of the active site, the imidazoline binding site, which motivated the present study.
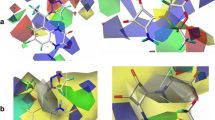
MAO-B is composed of three structural domains, as shown in Fig. 1. Briefly, the substrate domain is composed of residues Phe 103, Pro 104, Trp 119, Leu 164, Leu 167, Leu 171, Phe 168, Ile 199, Ile 316 and Tyr 326. The flavin domain is composed of the flavin group covalently bound to Cys397. There is also a transmembrane domain, composed by alpha helices. The substrate entrace is close to the intersection of the enzyme with mitocondrial surface [9, 31].

The crystal structure of human MAO-B. Structure of monoamine oxidase in complex with FAD and 2-(2-benzofuranyl)-2-imidazoline (PDB access code: 2XCG)
Molecular docking simulation (MDS) is a computational methodology that provides automatic means to determine the conformation of a proteins-ligand complex. Considering protein-ligand interactions, it is possible to visualize that this computer simulation is equivalent to the key-and-lock problem, where the lock is the protein and the key the ligand. The main objective of the MDS is to adjust the position of the ligand (key) in the protein (lock). In a typical MDS it generates many potential positions for the ligand in the protein, known as poses. Consequently, it is necessary to have a model, which will allow evaluation of all possible positions for the ligand, and then choose the best position. This model of selection could be expressed as an energy function [32] or a scoring function [33–38] not necessarily related to an energy function.
Here we applied molecular docking search engines and empirical scoring functions implemented in the program molegro virtual docker [39–41] to evaluate the interaction of MAO-B with ligands. The goals of the present paper are the following: 1) To establish a fast and reliable molecular docking protocol to identify ligand position in the imidazole-binding site on MAO-B. 2) To apply this docking protocol to predict ligand-binding affinity, and 3) to identify new potential MAO-B inhibitors, with focus on the imidazoline-binding site. We describe an optimized molecular docking protocol that was able to predict ligand position with RMSD lower than 0.3 Å when compared with the crystallographic structure. This docking protocol was able to predict inhibitory activity, further validating this docking protocol. Application of this reliable protocol was able to identify new potencial MAO-B inhibitors. Their intermolecular interactions and structural features are discussed.
Materials and methods
Re-docking and cross-docking
MVD [39–41] is one of the available commercial programs for docking simulations based on evolutionary algorithms. Recent evaluation of MVD strongly indicates that it is capable of superior overall performance when compared with AUTODOCK, SURFLEX, FLEXX and GOLD [39–42]. MVD brings implementation of four search algorithms to find ligand position and orientation. They are: MOLDOCK optimizer (implementation of differential evolution algorithm), MOLDOCK simplex evolution (implementation of downhill simplex method), iterated simplex, and iterated simplex (with ant colony optimization) [39–41].
In this study, prior to MDS, all atom types and the bond orders were corrected to both ligand and monoamine oxidase B structures using the MVD automatic preparation function [39]. For each complex, the hydrogen atoms were added and the MVD default partial charges were assigned. Compounds ZINC00154386 and ZINC00057128 are cationic molecules and ZINC02565373 is anionic. All others compounds are neutral. This automatic preparation of the structures was also applied to all ligands used in the test set and virtual screening (VS) (described below). Molecular cavities were detected using the grid-based cavity prediction algorithm. All water molecules were deleted from the original PDB files for re-docking and cross-docking (described below) simulations. Re-docking simulations of the ligand 2-BFI against the imidazoline binding site on MAO-B were carried out using the atomic coordinates of 2XCG [31].
In addition to re-docking, a procedure called "cross-docking" can also be used to further validate a docking protocol. Considering that several crystallographic structures are available for the same protein, cross-docking can be applied. This procedure involves docking a number of ligands found in a variety of crystal structures of a protein identical to a single rigid protein crystallographic conformation [43]. When a protein target presents major conformational changes upon ligand binding, a significant difference is expected between the crystallographic and docked structures. In the cross-docking simulations we used 10 crystallographic structures (PDB access codes: 1OJ9, 2VRL, 2VZ2, 2XFN, 2XFO, 2XFP, 2XFQ, 2XFU, 3PO7, 2VRM). Re-docking and cross-docking are the initial stages of all VS projects. The overall scheme is shown in Fig. 2, and it has been fully described elsewhere [44]. Briefly, phase 1 is focused on selection and validation of a docking protocol, as described above. Phase 1 ends when an adequate protocol is found (selection criterion RMSD < 2.0 Å). It should be pointed out that the RMSD criterion is dependent on the number of torsion angles, and a less demanding criterion may be adopted for re-docking of a ligand with a number of torsion angles higher than 10 [40, 41]. Once a docking protocol is chosen we select a small-molecule database to be used in the screening (phase 2). In phase 3, we start docking simulations for each ligand present in the selected database. During a typical docking simulation several orientations can be obtained for each ligand. Here we selected the one with the lowest scoring function. The scoring function used by MVD improves accuracy of scoring functions with a new hydrogen bonding term and new charge schemes. Four scoring functions are implemented in the MVD, including MOLDOCK score and PLANTS score [39]. These two functions offer grid-based versions, in which hydrogen bond directionality is not considered.

Flowchart of virtual screening process (modified from [44]). Phase 1 is focused on selection and validation of a docking protocol. Phase 2 is the selection of a small-molecule database to be used in the screening. In phase 3, we start docking simulations for each ligand present in the selected database. In phase 4, analysis of the best scored ligands are carried out
To further improve docking accuracy, a re-ranking scoring function was used. This function identifies the most promising docking solution from the solutions obtained by the docking algorithm [39]. Re-rank score includes the docking scoring function terms, such as a sp2-sp2 torsion term and a Lennard-Jones 12-6 potential. The re-ranking score function is computationally more expensive than the scoring functions (MOLDOCK and PLANTS scores) used during the docking simulation but it is usually better than the docking score function at determining the best pose among several poses originating from the same ligand and also for evaluating ligand-binding affinity. Furthermore, due to the stochastic nature of the search algorithm, we applied the same VS protocol 16 times, running the best docking protocol in 16 computers in parallel (coarse-grain parallelism) and only considered a compound as a hit if it is present in the majority of the VS results. In the present work, all simulations were performed on 16 iMac computers (Intel Processor Core 2 Duo, 2.66 GHz, 2 GB SDRAM DDR3 1066 MHz).
After identification of potential inhibitors by MDSs, the best scored ligands can be submitted to the web server FAF-Drugs [45] in order to assess physical-chemical properties (phase 4). These are key properties that need to be considered in early stages of the drug discovery process, and FAF-Drugs allows users to filter molecules via simple rules such as molecular weight, polar surface area, logP and number of rotatable bonds. The ligands were filtered following the Lipinski’s rule of five (RO5). RO5 advocates that drugs which present oral bioavailability, in general, follow: molecular weight less or equal to 500, LogP less or equal to 5, number of hydrogen bond donor groups less or equal to 5 and number of hydrogen bond acceptor groups less or equal to 10 [46].
To confirm the ability of the MVD program to evaluate the inhibitory activity of a ligand bound to MAO-B we selected 13 MAO-B inhibitors for which experimental information for their inhibition activity (IC50) was available. This information was retrieved from the BRENDA [47]. To be consistent in our comparisons, all data retrieved from BRENDA database were checked to confirm they all belong to the same biomolecular and expression systems (MAO-B). We used the program ACD/ChemSketch from Advanced Chemistry Development (Toronto, Canada)(http://www.acdlabs.com/products/draw_nom/) to generate inhibitor structures and submitted them to the automatic preparation of the structures of MVD (described above). From now on this ensemble of structures will be referred to as test set.
In order to identify new commercially available MAO-B inhibitors we focused our VS efforts on the SIGMA library. This small-molecule library presents 15,186 compounds. We downloaded these compounds in the structure-data file (SDF) format from the ZINC database [48–50]. The atomic coordinates for the structure 2XCG [31] was used as a target for VS and test set studies, water molecules and ligands were deleted from the structure. We used the best docking protocol with re-ranking score function identified in phase 1 to evaluate binding affinity.
Results and discussion
Re-docking and cross-docking
One of the most important results derived from several applications of MVD program is the comparison of re-docking simulations between MVD and other docking programs, such as GOLD, FLEXX, GLIDE, and AUTODOCK [39–42]. These comparisons strongly indicated that the MVD is able to obtain lower RMSDs in the great majority of the analyzed crystallographic structures (re-docking simulations). In addition, MVD not only presents a better overall performance in re-docking simulations, but it is also faster than AUTODOCK [42], one of the first MDS program. These results demonstrate that MVD is reliable; therefore, we used it in the present work.
Initially, a search for the best molecular docking protocol was performed. The structure of 2-BFI in complex with tranylcypromine-inhibited MAO-B was used for re-docking simulations. The key criterion describing the quality of a MDS is the RMSD. In molecular docking applications, the best binary complex is the one closer to the structure determined by x-ray crystallography. Analysis of the re-docking results for the combination of four search algorithms and four scoring functions (a total of 16 different docking protocols) generated RMSD from 0.2 to 12.08 Å. Table 1 shows the RMDS for all docking protocols. The best results were obtained for the following search engines: MOLDOCK SE, MOLDOCK optimizer, and iterated simplex. Iterated simplex with ant colony optimization generated the worst RMSD (12.08 Å). The parameters for docking, especially the search engine features, were optimized by running several MDSs on the complex structure. The following parameters and their combinations were varied: radius of the docking sphere, number of runs, maximum number of iterations, and maximum population size. The optimized parameters for the docking are the following:
-
Scoring function
-
Empirical scoring function: Re-rank score (used for ranking the MDS results)
-
-
Binding site
-
Origin: x = 50.63; y = 161.17 and z = 31.34 Å
-
Radius: 10 Å
-
-
Search algorithm
-
Algorithm: MOLDOCK SE
-
Number of runs: 10
-
Constrain poses to cavity: Enabled
-
-
Parameter settings
-
Max iterations: 1500
-
Max population size = 50
-
-
Pose generation
-
Energy threshold: 100.00
-
-
Simplex evolution
-
Max steps: 300
-
Neighbor distance factor: 1.00 .
-
Figure 3 shows the docking sphere used in the re-docking simulations. Since the combination of MOLDOCK SE and re-rank score, generated very low RMSD (0.2 Å) we chose this docking protocol and used it in all further MDSs. Cross-docking simulations for 10 MAO-B complexes using this protocol generated RMDS from 1.45 to 1.80 Å, further validating the present docking protocol.

Search space sphere (green) defined for molecular docking simulations
Relationships between the MAO-B inhibition and re-rank score
Predicting the ligand-binding affinity based on a static conformation of the ligand is a complex task. For example, energetic contributions from solvent interactions and entropy contributions are complicated to handle in the simplified models used in MDS. While the re-rank score in MVD provides an approximation of the potency of the intermolecular interaction, it is not calibrated in physical-chemical units and it does not take intricate contributions (such as entropy) into account. Even though the re-rank score might be successful in ranking different poses of the same ligand, it might be less successful in ranking poses of different ligands. So, to test whether re-ranking score is able to predict ligand-binding affinity we applied the best docking protocol to a test set of 13 ligands for which experimental information was available.
It is expected that scoring functions show a correlation with inhibitory activity (IC50). To carry out comparisons we used pIC50, which is the log (IC50), as shown on Table 2. Since there is no direct significant relationship between the two datasets, we have to calculate the Spearman’s rank order correlation coefficient to evaluate the statistical significance of the relationship between these two independent variables (re-rank score and pIC50). The equation for this correlation coefficient is as follows,
where N is the number of pairs, 13 in our case, the variables r(xj) and r(yj) are the rank of the pIC50 and the re-rank score of the jth sample in the dataset [22].
Analysis of the Spearman’s rank correlation coefficient for this dataset (Table 2) generated a ρ = 0.8, which is higher than the critical value at the 0.002 level of significance. It implies that the re-rank score of MVD can be applied for activity prediction.
Furthermore, application of the best docking protocol to simulate the interaction of 2-BFI with the structure of Ile199Ala mutant of human MAO B (PDB access code: 2XFO) generated a re-rank score of −72.06, higher than the re-rerank for 2-BFI in complex with tranylcypromine-inhibited MAO-B (-86.57). Previously published study [31] indicated that 2-BFI binds to the mutant enzyme weakly with a Ki of approximately 58 μM, whereas 2-BFI binds to tranylcypromine inhibited human MAO B with a Kd value of 9 nM, which is nearly 1000-fold increase in binding affinity for 2- BFI on tranylcypromine inhibition specifically observed in human MAO B. These experimental results are in agreement with re-rank scores estimated with MVD. Taken together these results indicate that this docking protocol can also be applied to evaluate the binding-affinity of compounds identified in a VS study.
Virtual screening
VS is a computational approach used to identify potential new inhibitors for a protein target for which the 3D structure is available. It has been successfully applied to identify a plethora of inhibitors [40, 41, 50]. Our focus here is on the identification of new potential inhibitors for MAO-B. Although previous VS studies have been perfomed on the MAO-B [13–15]. There is no docking studies using the combination of simplex evolution search algorithm and MOLDOCK score. Furthermore, this is the first VS study focused on the imidazoline-binding site on MAO-B.
Application of the previously described docking protocol to a database with 15,186 compounds retuned as best results 11 compounds, with re-rank score below −100. Among these 11 compounds, two stand out, since they were found among the best hits in 14 out 16 simulations. They are: ZINC00154386 ( 3-(1,3-benzodioxol-5-yl)-5-piperidin-1-ium-4-yl-1,2,4-oxadiazole ) and ZINC02387301 (5-[5-(5-formylthiophen-2-yl)thiophen-2-yl]thiophene-2-carbaldehyde)).
We used filtering options of FAF-Drugs [45] to this set of 11 compounds, based on Lipinski’s rules [46]. A total of eight compounds passed to this filter (Table 3) (Fig. 4), among them the molecules ZINC00154386 and ZINC02387301. Re-rank scores for these eight compounds range from −116.958 to −100.029.

Molecular structures of the top-scoring compounds identified in the VS protocol. a ZINC02387301. b ZINC00154386. c ZINC02169849. d ZINC00644889. e ZINC00056610 f ZINC02565373 g ZINC01724292 h ZINC00057128
Intermolecular interactions
In order to assess intermolecular contacts between the compounds identified in the VS and the MAO-B we used the program LIGPLOT [51]. Analysis of the intermolecular interaction indicates key residues responsible for ligand binding specificity. Intermolecular hydrogen bonds involving residues Tyr326 and Pro102. Van der Waals contacts are present between the ligand and the residues Phe 168, Leu 164, Il 316, Il 199, Leu 171, and Leu 88. The importance of the residues Leu88, Pro102 and Leu164 for intermolecular interactions has been highlighted in a recent study [52] where molecular dynamics simulations of MAO B in a lipid bilayer were carried out. Theses simulations indicated that the bilayer controls the accessibility of the imidazoline-binding domain on MAO-B by the movement of two key loops that form the active site entrance (residues 85–110 and 155–165). Furthermore, the molecular dynamics simulation indicated the stability of the imidazoline-binding domain on MAO-B [52]. Table 4 shows intermolecular interactions for all eight compounds identified in the VS. There are no van der Waals contacts closer than the sum of their van der Waals radii, and the intermolecular hydrogen bonds follow the criteria established in the LIGPLOT algorithm [51]. All eight compounds present interactions with residues Ile316, Ile199 and Leu88. Out of eight compounds, six show intermolecular interactions with residues Leu171, Leu167, and Leu171. The residue Tyr326 is found in interactions with ligands 1, 2, 3, 4, 5, 6 and 7, which strongly indicates the importance of these residues for ligand specificity, as has been suggested from the analysis of the crystallographic structure of 2-BFI in complex with tranylcypromine-inhibited MAO-B [31].
Conclusions
We have used simplex evolution algorithm to carry out flexible docking search for prediction of MAO-B inhibitory activities. The parameters for flexible docking were optimized to allow us routine work. After that, cross-docking simulations were performed with the imidazole-binding site. We have calculated the rank correlation between the MOLDOCK score of the program MVD and the respective MAO-B inhibitory activity. The calculated correlation coefficients imply that the docking using the combination of simplex evolution algorithm and MOLDOCK score is a suitable technique for making qualitative predictions about activity. At the same time, the results confirm the assumption that there is a considerable relationship between the IC50 and re-rank scores. All experimental information used in comparison with scoring functions followed Michaelis–Menten equation which describes a unireactant process in which velocity is related to substrate concentration in either a hyperbolic or linear way. A recent publication [53] discusses a novel analytical approach that offers a significant improvement upon the use of the Michaelis–Menten equation for analysis of MAO kinetic data. Nevertheless, our correlation analysis was based upon the classical model due to the reduced number of MAO-B inhibitors (three inhibitors) investigated in this novel analytical approach [53], which makes statistical analysis (Spearman’s rank order correlation coefficient) difficult.
MAOs are protein targets for development of drugs to treat many neuropsychiatric and neurodegenerative conditions [9, 53, 54]. IMAOs are used in the treatment of PD focused on MAO-B in particular (as a result affecting dopaminergic neurons), as well as providing a substitute for migraine prophylaxis [54]. Therefore, the development of a new generation of MAO-B inhibitors is of great interest. We have studied here the influence of key residues on ligand interaction with MAO-B and attempted to predict new possibly active compounds. We have identified two compounds (ZINC00154386 and ZINC02387301) with low re-rank score. Both compounds present intermolecular interactions involving residues Tyr326 and Pro102, as observed for the crystallographic structure 2XCG [31]. Information obtained in this study will be used for designing new MAO-B inhibitors and for additional work in the area of molecular docking simulations.
Abbreviations
- PD:
-
Parkinson's disease
- IMAO:
-
Inhibitor of monoamine oxidase
- MAO:
-
Monoamine oxidase
- MAO-A:
-
Monoamine oxidase A
- MAO-B:
-
Monoamine Oxidase B
- FAD:
-
Flavin –adenine - dinucleotide
- VS:
-
Virtual screening
- 2-BFI:
-
2-(2-benzofuranyl)-2-imidazoline
- RMSD:
-
Root mean square deviation
- PDB:
-
Protein data bank
- EA:
-
Evolutionary algorithm
- PLP:
-
Piecewise linear potencial
- E intermol:
-
Energia intermolecular
- E intramol:
-
energia intramolecular
- RO5:
-
Lipinski’s rule of five
- ρ:
-
Coeficiente de Spearman
- IUPAC:
-
International Union of Pure and Applied Chemistry
- IC50 :
-
Half maximal inhibitory concentration
References
Naoi M, Maruyama W (2010) Monoamine oxidase inhibitors as neuroprotective agents in age-dependent neurodegenerative disorders. Curr Pharm Des 16(25):2799–817
Naoi M, Maruyama W, Akao Y, Yi H, Yamaoka Y (2006) Involvement of type A monoamine oxidase in neurodegeneration: regulation of mitochondrial signaling leading to cell death or neuroprotection. J Neural Transm Suppl 71:67–77
Edmondson DE, Binda C, Mattevi A (2007) Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Arch Biochem Biophys 464(2):269–276
Shih JC, Chen K, Ridd MJ (1999) Monoamine oxidase: from genes to behavior. Annu Rev Neurosci 22:197–217
Birkmayer W, Knoll J, Riederer P, Youdim MB, Hars V, Marton J (1985) Increased life expectancy resulting from addition of L-deprenyl to Madopar treatment in Parkinson’s disease; a long-term study. J Neural Transm 64(2):113–127
Edmondson DE, Mattevi A, Binda C, Li M, Hubálek F (2004) Structure and mechanism of monoamine oxidase. Curr Med Chem 11(15):1983–1993
Riederer P, Konradi C, Schay V, Kienzl E, Birkmayer G, Danielczyk W, Sofic E, Youdim MB (1987) Localization of MAO-A and MAO-B in human brain: a step in understanding the therapeutic action of L-deprenyl. Adv Neurol 45:111–118
Youdim MBH, Bakhle YS (2006) Monoamine oxidase: isoforms and inhibitors in Parkinson’s disease and depressive illness. Br J Pharmacol 147(Suppl 1):S287–296
Youdim MB, Edmondson D, Tipton KF (2006) The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci 7(4):295–309
Riederer P, Lachenmayer L, Laux G (2004) Clinical applications of MAO-inhibitors. Curr Med Chem 11(15):2033–2043
Manley-King CI, Bergh JJ, Petzer JP (2011) Inhibition of monoamine oxidase by C5-substituted phthalimide analogues. Bioorg Med Chem 19(16):4829–4840
Maccioni E, Alcaro S, Cirilli R, Vigo S, Cardia MC, Sanna ML, Meleddu R, Yanez M, Costa G, Casu L, Matyus P, Distinto S (2011) 3-Acetyl-2,5-diaryl-2,3-dihydro-1,3,4-oxadiazoles: a new scaffold for the selective inhibition of monoamine oxidase B. J Med Chem 54(18):6394–6398
Gaspar A, Silva T, Yáñez M, Vina D, Orallo F, Ortuso F, Uriarte E, Alcaro S, Borges F (2011) Chromone, a privileged scaffold for the development of monoamine oxidase inhibitors. J Med Chem 54(14):5165–5173
Gaspar A, Teixeira F, Uriarte E, Milhazes N, Melo A, Cordeiro MN, Ortuso F, Alcaro S, Borges F (2011) Towards the discovery of a novel class of monoamine oxidase inhibitors: structure-property-activity and docking studies on chromone amides. Chem Med Chem 6(4):628–632
Shelke SM, Bhosale SH, Dash RC, Suryawanshi MR, Mahadik KR (2011) Exploration of new scaffolds as potential MAO-A inhibitors using pharmacophore and 3D-QSAR based in silico screening. Bioorg Med Chem Lett 21(8):2419–2424
Delogu G, Picciau C, Ferino G, Quezada E, Podda G, Uriarte E, Viña D (2011) Synthesis, human monoamine oxidase inhibitory activity and molecular docking studies of 3-heteroarylcoumarin derivatives. Eur J Med Chem 46(4):1147–1152
Reniers J, Robert S, Frederick R, Masereel B, Vincent S, Wouters J (2011) Synthesis and evaluation of ß-carboline derivatives as potential monoamine oxidase inhibitors. Bioorg Med Chem 19(1):134–144
Jia Z, Zhu Q (2010) 'Click' assembly of selective inhibitors for MAO-A. Bioorg Med Chem Lett 20(21):6222–6225
Khattab SN, Hassan SY, Bekhit AA, El Massry AM, Langer V, Amer A (2010) Synthesis of new series of quinoxaline based MAO-inhibitors and docking studies. Eur J Med Chem 45(10):4479–4489
Maccioni E, Alcaro S, Orallo F, Cardia MC, Distinto S, Costa G, Yanez M, Sanna ML, Vigo S, Meleddu R, Secci D (2010) Synthesis of new 3-aryl-4,5-dihydropyrazole-1-carbothioamide derivatives. An investigation on their ability to inhibit monoamine oxidase. Eur J Med Chem 45(10):4490–4498
Geldenhuys WJ, Darvesh AS, Funk MO, Van der Schyf CJ, Carroll RT (2010) Identification of novel monoamine oxidase B inhibitors by structure-based virtual screening. Bioorg Med Chem Lett 20(17):5295–5298
Parini A, Moudanos CG, Pizzinat N, Lanier SM (1996) The elusive family of imidazoline binding sites. Trends Pharmacol Sci 17(1):13–16
Raddatz R, Parini A, Lanier SM (1997) Localization of the imidazoline binding domain on monoamine oxidase B. Mol Pharmacol 52(4):549–553
Lanier SM, Lanier B, Bakthavachalam V, McGrath CR, Neumeyer JL (1995) Use of high affinity, radioiodinated probes for identification of imidazoline/guanidinium receptive sites. Ann NY Acad Sci 763:106–111
Remaury A, Raddatz R, Ordener C, Savic S, Shih JC, Chen K, Seif I, De Maeyer E, Lanier SM, Parini A (2000) Analysis of the pharmacological and molecular heterogeneity of I(2)-imidazoline-binding proteins using monoamine oxidase-deficient mouse models. Mol Pharmacol 58(5):1085–1090
Anderson NJ, Seif I, Nutt DJ, Hudson AL, Robinson ES (2006) Autoradiographical distribution of imidazoline binding sites in monoamine oxidase A deficient mice. J Neurochem 96(6):1551–1559
Ozaita A, Olmos G, Boronat MA, Lizcano JM, Unzeta M, Garcia-Sevilla JA (1997) Inhibitons of MAO A and B activities by imidazol(ine)/guanidine receptive drugs, nature of the interaction and distinction from I2-imidazoline receptors in rat liver. Br J Pharmacol 121(5):901–912
Jones TZ, Giurato L, Guccione S, Ramsay RR (2007) Interactions of imidazoline ligands with the active site of purified monoamine oxidase A. FEBS J 274(6):1567–1575
Raddatz R, Parini A, Lanier SM (1995) Imidazoline/guanidinium binding domains on monoamine oxidases. Relationship to subtypes of imidazoline-binding proteins and tissue-specific interaction of imidazoline ligands with monoamine oxidase B. J Biol Chem 270(46):27961–27968
Raddatz R, Savic SL, Bakthavachalam V, Lesnick J, Jasper JR, McGrath CR, Parini A, Lanier SM (2000) Imidazoline-binding domains on monoamine oxidase B and subpopulations of enzyme. J Pharmacol Exp Ther 292(3):1135–1145
Bonivento D, Milczek EM, McDonald GR, Binda C, Holt A, Edmondson DE, Mattevi A (2010) Potentiation of ligand binding through cooperative effects in monoamine oxidase B. J Biol Chem 285(47):36849–36856
Klebe G, Böhm HJ (1997) Energetic and entropic factors determining binding affinity in protein-ligand complexes. J Recept Signal Transduct Res 17(1–3):459–473
Böhm HJ (1994) The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. J Comput Aided Mol Des 8(3):243–256
Böhm HJ (1996) Current computational tools for de novo ligand design. Curr Opin Biotechnol 7(4):433–436
Böhm HJ (1998) Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J Comput Aided Mol Des 12(4):309–323
Stahl M, Böhm HJ (1998) Development of filter functions for protein-ligand docking. J Mol Graph Model 16(3):121–132
Wang R, Lai L, Wang S (2002) Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput Aided Mol Des 16(1):11–26
De Azevedo WF, Jr DR (2008) Computational methods for calculation ligand-binding affinity. Curr Drug Targets 9(12):1031–1039
Thomsen R, Christensen MH (2006) MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49(11):3315–3321
Heberlé G, De Azevedo WF Jr (2011) Bio-inspired algorithms applied to molecular docking simulations. Curr Med Chem 18(9):1339–1352
De Azevedo WF Jr (2010) MolDock applied to structure-based virtual screening. Curr Drug Targets 11(3):327–334
Araújo JQ, Lima JA, Pinto Ada C, de Alencastro RB, Albuquerque MG (2011) Docking of the alkaloid geissospermine into acetylcholinesterase: a natural scaffold targeting the treatment of Alzheimer's disease. J Mol Model 17(6):1401–1412
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shaw DE, Shelley M, Perry JK, Francis P, Shenkin PS (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47(7):1739–1749
Vianna CP, de Azevedo WF Jr (2012) Identification of new potential Mycobacterium tuberculosis shikimate kinase inhibitors through molecular docking simulations. J Mol Model 18(2):755–764
Miteva MA, Violas S, Montes M, Gomez D, Tuffery P, Villoutreix BO (2006) FAF-drugs: free ADME/tox filtering of compound collections. Nucleic Acids Res 34:W738–W744
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46(1–3):3–26
Scheer M, Grote A, Chang A, Schomburg I, Munaretto C, Rother M, Söhngen C, Stelzer M, Thiele J, Schomburg D (2011) BRENDA, the enzyme information system in 2011. Nucleic Acids Res 39:D670–6
Irwin JJ, Shoichet BK (2005) ZINC - a free database of commercially available compounds for virtual screening. J Chem Inf Model 45(1):177–182
Timmers LF, Pauli I, Caceres RA, De Azevedo WF Jr (2008) Drug-binding databases. Curr Drug Targets 9(12):1092–1099
De Azevedo WF Jr (2010) Structure-based virtual screening. Curr Drug Targets 11(3):261–263
Wallace AC, Laskowski RA, Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng 8(2):127–134
Allen WJ, Bevan DR (2011) Steered molecular dynamics simulations reveal important mechanisms in reversible monoamine oxidase B inhibition. Biochemistry 50(29):6441–6454
Ramsay RR, Olivieri A, Holt A (2011) An improved approach to steady-state analysis of monoamine oxidases. J Neural Transm 118(7):1003–1019
Dowson JH (1987) MAO inhibitors in mental disease: their current status. J Neural Transm Suppl 23:121–138
Acknowledgments
This work was supported by grants from the National Council for Scientific and Technological Development of Brazil (CNPq) and Instituto Nacional de Ciência e Tecnologia do Conselho Nacional de Desenvolvimento Científico e Tecnológico-Ministério de Ciência e Tecnologia (INCT-Tuberculose, CNPq-MCT, Brazil). WFA is senior researcher for CNPq (Brazil). The fellowship from CNPq-Brazil is also acknowledged (FPM).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moraes, F.P., de Azevedo, W.F. Targeting imidazoline site on monoamine oxidase B through molecular docking simulations. J Mol Model 18, 3877–3886 (2012). https://doi.org/10.1007/s00894-012-1390-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-012-1390-7