
Abstract
Studies of the the three-dimensional quantitative structure–activity relationships for ninety-five c-kit tyrosine kinase inhibitors were performed. Based on a co-crystallized compound (1 T46), known inhibitors were aligned with c-kit by induced-fit docking, and multiple training/test set splitting was performed to validate the selected pharmacophore model. The best pharmacophore model consisted of five features: one hydrogen-bond donor and four aromatic rings. Reliable statistics were obtained (R 2 = 0.95, R 2pred = 0.75), and the model was validated by using it to select c-kit inhibitors from a database; 82.1% of the hits it retrieved were active. Accordingly, our model can be reliably used to identify new c-kit inhibitors, and can provide useful information when designing new inhibitors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The c-kit proto-oncogene encodes a transmembrane tyrosine kinase receptor that is activated by the stem cell factor (SCF), its natural ligand. C-kit protein plays a critical role in modulating histamine release from mast cells [1, 2] following its binding with SCF, which leads to dimerization and autophosphorylation at specific tyrosine residues. Moreover, signaling by c-kit plays an important role in cellular transformation and differentiation, including proliferation, survival, adhesion, and chemotaxis [3]. The overexpression of the c-kit proto-oncogene has been reported in hematopoietic cells, small cell lung cancer, and gastrointestinal stromal tumors [4–6]. Furthermore, it has been demonstrated that mutations of c-kit protect human colon adenocarcinoma cells against apoptosis and enhance their invasive potential [7]. The clinical importance of c-kit expression in tumors has led to research focused on finding inhibitors of this tyrosine kinase. Imatinib (Gleevec®) was the first such compound to be used in therapy, but mutations of c-kit led to reduced efficacy or a complete lack of efficacy of this treatment. Other compounds are likely to be effective against mutants, such as sunitinib (Sutent®), but the need for new and more effective inhibitors is still critical. In this paper, we report a three-dimensional quantitative structure–activity relationship (3D-QSAR) analysis of 95 known c-kit inhibitors that were initially docked into the crystal structure of c-kit by means of a mixed approach including molecular dynamics and docking (induced fit). The model obtained revealed interesting features that should be considered during the design and development of new potentially active candidates targeting this kinase, which could be useful as anticancer agents.
Materials and methods
Dataset for analysis
A dataset of 95 compounds (2-aminobenzoxazole derivatives [8], 3-aminobenzoisoxa(thi)azole derivatives [9, 10], thienopyrimidine derivatives [11], and anilinophthalazine derivatives [12]) was selected for the present study (Table 1). Their pIC50 values were used as a dependent variable in the QSAR models. In order to validate each model, the inhibitors were split into a training set and a test set. Two different splitting methods were applied. The first method involved automated random selection, while the second involved a Kohonen map artificial neural network (ANN) or self-organizing maps (SOM) [13, 14]. Due to their clustering capabilities, Kohonen maps ensure that both sets are homogeneously distributed within the entire area of descriptor space. The test compounds were selected by evaluating the minimum distance from the centroid of each cell in the top map. Selection in this manner allows predictions to be made by interpolation and not extrapolation from the domain of the particular QSAR model [15]. Descriptor calculations were carried out using the CODESSA software package [16], and descriptor space was explored using Kohonen maps for autoscaled data. This structural information was used to build a Kohonen map (five per five neurons, 300 epochs) [17]. After 300 epochs of net training, similar compounds are clustered together in the multi-dimensional descriptor space. In each splitting method, 76 of the 95 inhibitors were chosen for a training set and 19 were selected as a test set (Table 1).
Ligand preparation
The 95 ligands were processed with the LigPrep software package in order to assign the appropriate protonation states to them at physiological pH (7.2 ± 0.2), employing the Ionizer option. Conformers were generated through MacroModel torsional sampling using the OPLS_2005 force field [18].
Induced-fit docking
Generated conformers were docked into the c-kit crystallographic structure (PDB: 1 T46), which was originally complexed with imatinib, and the best score poses for each ligand were used to generate pharmacophore hypotheses. The mixed molecular docking/dynamics protocol called induced-fit docking (IFD) [19] was used. In an interative manner, this approach combines ligand-docking techniques with those used to model receptor conformational changes. The Glide docking software package [20] was used for ligand flexibility, while the refinement module in the Prime program [21] was used to account for receptor flexibility: the degrees of freedom of side chains were mainly sampled, while minor backbone movements were allowed through minimization. The strategy used was to first dock ligands into a rigid receptor using a softened energy function such that steric clashes do not prevent at least one pose from assuming a conformation close to the correct one (the “ligand sampling step”). The degrees of freedom of the receptor were then sampled, and global ligand/receptor energy minimization was performed for many ligand poses; this attempted to identify low free-energy conformations of the whole complex (the “protein sampling step”). After that, a second ligand docking step was performed on the refined protein structures, using a hard potential function to sample the ligand’s conformational space within the refined protein environment (the “ligand resampling step”). Finally, a composite score function was applied to rank the complexes; this accounted for the receptor/ligand interaction energy as well as strain and solvation energies (the “scoring step”). The composite score, which was used to perform the final ranking of the compounds, was derived as follows:
3D-QSAR pharmacophore modeling
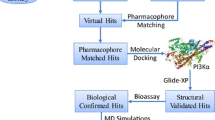
The 3D-QSAR study was performed using the Phase software package [22]. Phase utilizes fine-grained conformational sampling and a range of scoring techniques to identify common pharmacophore hypotheses. These convey characteristics of 3D chemical structures that are reported to be critical for binding. The pharmacophore model was developed by using a set of pharmacophore features to generate sites for all of the compounds. Each structure was represented by a set of points in 3D space that coincided with various chemical features which facilitated noncovalent binding between the ligand and its binding pocket. Phase provides a standard set of six pharmacophore features: hydrogen-bond acceptor (A), hydrogen-bond donor (D), hydrophobic group (H), negatively ionizable (N), positively ionizable (P), and aromatic ring (R). Hypotheses were generated by systematically varying the number of sites (n sites) and the number of matching active compounds (n act). With n act = n act_tot initially (n act_tot is the total number of active compounds in the training set), n sites was decreased from its initial value of 7 until at least one hypothesis was found and scored successfully. In this study, with n sites = 5 and n act = n act_tot = 8, common pharmacophores were examined using a scoring protocol to identify the pharmacophore from each surviving n-dimensional box that yielded the best alignment of the active-set ligands. The scoring protocol allows the different hypotheses to be ranked so that the most appropriate can be chosen for further investigation. Inactive molecules were also scored, in order to observe the alignment of these molecules with the pharmacophore hypotheses and to select the best ones. The larger the difference between the scores of the actives and inactives, the better the hypothesis is at discriminating active from inactive molecules. For QSAR development, models of the pharmacophore features of training-set molecules were placed into a regular grid of cubes, with each cube allotted zero or more “bits” to account for the different types of pharmacophore features in the training-set molecule that occupy the cube (1 Å). This representation gave rise to binary-valued occupation patterns that could be used as independent variables to create partial least-squares (PLS) factor 3D-QSAR models. Statistics on the correlation of the predicted with the actual activity data were collated for the hypothesis. Phase QSAR models can be either atom-based or pharmacophore-based, the difference being whether all atoms are taken into account, or whether only the pharmacophore sites that can be matched to the hypothesis are considered. The choice of model depends largely on whether or not the training-set molecules are sufficiently rigid and congeneric. If the structures contain a small number of rotatable bonds and have some of their structural framework in common, then an atom-based model may work quite well [22]. The selected dataset did not have many rotatable bonds, so the atom-based QSAR model was used.
Results and discussion
We started our work by redocking imatinib into the protein via the IFD approach in order to evaluate the reliability of the computational algorithm before carrying out receptor-guided alignment of the inhibitors. Training alignment of imatinib gave good results in terms of both IF score (crystallized score = −12.28; docked = −12.51), descriptor contributions (crystallized: Hbond = −1.5, vdW = −66.1, Coul = −9.1; docked: Hbond = −1.4, vdW = −66.8, Coul = −9.1), and root mean square deviation (RMSD = 0.43) (Fig. 1). Thus, it was possible to align the selected inhibitors using the same approach. The top-ranked binding poses, selected by IFD score and by visual inspection, were used to generate the pharmacophore hypotheses (Fig. 2). The 3D-QSAR studies were carried out using the Phase package from Schrödinger LLC. To find the common pharmacophore hypothesis, the dataset was split into an active set and an inactive set (the “pharm” set) (Table 1). Compounds with pIC50 > 8.00 were considered to be active, those with pIC50 < 6.00 were considered to be inactive, whereas those in-between were considered to be moderately active. Three hundred sixty-one hypotheses were identified (Table 2). Generated hypotheses were submitted to a Phase scoring procedure consisting of three scores: survival was calculated solely on the basis of the active set; surv-inactive was calculated on the basis of the active and inactive sets; post-hoc was calculated on the basis of the active and inactive sets, with a reward assigned based on the pIC50 of each compound of the data set. When a post-hoc score of >6.00 was taken to be the cut-off value, 68 hypotheses survived and were then used in the generation of QSAR models (Table 3). All molecules in the dataset were then aligned, matching to at least three pharmacophore features. Two different approaches to splitting the dataset into training and test sets were used: in the first, the compounds were randomly divided into a 76-member training set and 19-member test set, biasing both sets in order to give structural diversity to them both, and employing the standard 4:1 training/test ratio for a QSAR study. The other approach was based on a Kohonen map artificial neural network or SOM (Fig. 3). Descriptor calculations and autoscaling of the descriptor matrix were the starting point when performing the Kohonen clustering approach. The selected test set members were characterized by the minimum distance from the centroid of each cell in the top map. The standard ratio (76/19 sets) was used in this approach too.

Superposition of co-crystallized imatinib (yellow) and IFD (red)

Docked structures of the most active compounds

Kohonen top map
Both approaches to test-set selection selected the same 3D pharmacophore model, so this model was the only one evaluated any further. This best model was DRRRR.115, in which all active molecules in the active set matched the hypothesis.
The Phase statistical analysis for each test-set selection method is shown in Table 4. A statistical analysis that included the R 2 versus RMSE/SD plot was employed to choose the best PLS model for each set selection method. The best model was chosen on the basis of the PLS factor model’s minimum RMSE/SD value, where the R 2 value was still higher than 0.9. The principle of the 5:1 training set/PLS factor ratio was respected. The validity of each model was tested based on the calculated correlation coefficient for the test set. The squared correlation (random selection R 2pred = 0.72), the Pearson R (= 0.88), and the root mean square error for test set predictions (RMSE = 0.42) all confirm the good predictive capabilities of the final QSAR model for the test set. In the case of the Kohonen map ANN, the model with five PLS factors was chosen as the optimum one. The R 2pred (= 0.75), Pearson R (= 0.87), and RMSE (= 0.44) values confirm the validity of this model (Table 5; Fig. 4).

Actual versus predicted values for training and test sets: a random, b Kohonen
Analysis of the atom-based 3D-QSAR model
The best model, DRRRR.115, consists of four aromatic rings and one hydrogen-bond donor. The spatial arrangement of the pharmacophore sites shows that three aromatic rings, two of which are very close to each other, occupy the hydrophobic pocket created by the residues Val603, Leu595, Leu799, Phe811, and Tyr672. The hydrogen-bond donor site is located between the three aromatic rings and the fourth one, which is 8–10 Å away from the others (Fig. 5). The docking modes of the most active inhibitors resemble the docking pose of imatinib. In fact, for both the thienopyrimidines and aminobenzoisoxazoles, fused aromatic rings lie in the hydrophobic pocket created by the residues mentioned above. It should be emphasized that in each case the interactions of the target with the inhibitor are stabilized by at least two H-bonds. While the residues involved are Asp810, Cys673, and Thr670 in the case of imatinib, Glu640 and Glu671 are involved in the inhibitors from the dataset. Only in the case of compound 71 does the number of H-bonds with Cys673 rise up to four. For derivative 95, the residues involved are the same as those for imatinib. The substantial difference between imatinib and the inhibitors from the dataset is that the former acts as an H-acceptor for two sites and as an H-donor for one site, while the inhibitors from the dataset have only donor sites. It should be underlined that, although just one H-bond donor region and four aromatic features are present in the best model (DRRRR.115), analysis of the docking modes of the most active inhibitors indicates the formation of more extensive interaction patterns. However, in the pharmacophore hypothesis, the only features common to all active inhibitors in the data set are already taken into account. This means also that, for example, several compounds present more than four aromatic rings, but only four are common to all inhibitors. Figure 6 shows the volume occlusion maps for the atom-based 3D-QSAR model (donor, aromatic ring, electron-withdrawing features). These maps represent the regions of favorable and unfavorable interactions (shown in blue and red, respectively). The volume occlusion map for the H-bond donor describes the favorable 3D arrangement of hydrogen-bonding interactions with acceptor groups of the protein. The occlusion map surrounding the active molecules shows blue cubes opposite the oxazole or pyrimidine ring, which describe a favorable H-bond with an acceptor group of the protein.

Pharmacophore mapping of the most active compounds (left); superposition of active compounds on the pharmacophore

Occlusion maps. Left: hydrophobic; middle: donor; right: electron-withdrawing features
The volume occlusion map for the electron-withdrawing groups indicates that the most suitable position for this kind of group is in the external aromatic ring, while the presence of these groups in the inner aromatic ring appears to be unfavorable. This analysis indicates that improvements in binding affinity can be achieved by adding electron-withdrawing groups to the external aromatic ring. The hydrophobic volume occlusion map shows mixed coloration, indicating that an increase in the activity can be expected if the marked hydrophobicity of inhibitors is balanced by the presence of hydrophilic features or a reduction in the hydrophobicity.
The model was further validated by assessing its ability to pick out c-kit inhibitors in a known database aside from the model dataset, using the goodness-of-hit (GH score) approach [23]. For this external validation, a database of 234 compounds was created which included 34 known c-kit inhibitors, the structures of which are provided in the “Electronic supplementary material.” The structures of the known inhibitors were taken from the binding database. The other compounds employed in the test set used for external validation were selected from among the structures included in the ZINC database, with care taken to ensure that none of the molecules was structurally correlated with those in the original training and test sets. A summary of the results is provided in Table 6. When the 3D-QSAR model DRRRR.115 was used to query this database, 28 molecules were retrieved as hits (H t); among these, 23 molecules were known active inhibitors (H a). The calculated GH score and enrichment factor for the model were 0.74 and 4.83, respectively. The numbers of false positives and false negatives were 11 and 5, respectively. Thus, 82.14% of the hits retried by the model from the database were active inhibitors.
Conclusions
In summary, the goal of this study was to establish a strong relationship between structural features and inhibitory activity. Using a selected set of c-kit inhibitors, a 3D-QSAR pharmacophore model was obtained, consisting of one donor site and four aromatic rings. Volume occlusion maps demonstrated that inhibitory activity can be increased by modulating the donor abilities of the nitrogen or oxygen atoms in the fused aromatic rings that are involved in the H-bond interactions with the binding site of the receptor. This model was validated by predicting the active inhibitors in a test set prediction, and then by using it to pick active inhibitors from a known database. The model generated can be used to query databases and to provide guidelines when designing more potent inhibitors.
References
Eklund KK (2007) Mast cells in the pathogenesis of rheumatic diseases and as potential targets for anti-rheumatic therapy. Immunol Rev 217:38–52
Marshall J (2004) Mast-cell responses to pathogens. Nat Rev Immunol 4:787–799
Linnekin D (1999) Early signaling pathways activated by c-kit in hematopoietic cells. Int J Biochem Cell Biol 31:1053–1074
Hirota S, Isozaki K, Moriyama Y et al (1998) Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science (Washington, DC) 279:577–580
Wang WL, Healy ME (2000) Growth inhibition and modulation of kinase pathways of small cell lung cancer cell lines by the novel tyrosine kinase inhibitor STI 571. Oncogene 19:3521–3528
Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ (2000) Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 96:925–932
Bellone G, Carbone A, Sibona N et al (2001) Aberrant activation of c-kit protects colon carcinoma cells against apoptosis and enhances their invasive potential. Cancer Res 61:2200–2206
Potashman MH, Bready J et al (2007) Design, synthesis, and evaluation of orally active benzimidazoles and benzoxazoles as vascular endothelial growth factor-2 receptor tyrosine kinase inhibitors. J Med Chem 50:4351–4373
Kunz RK, Rumfelt S (2008) Discovery of amido-benzisoxazoles as potent c-kit inhibitors. Bioorg Med Chem Lett 18:5115–5117
Ji Z, Ahmed AA, Albert DH et al (2008) 3-Amino-benzo[d]isoxazoles as novel multitargeted inhibitors of receptor tyrosine kinases. J Med Chem 51:1231–1241
Dai Y, Guo Y, Frey RR et al (2005) Thienopyrimidine ureas as novel and potent multitargeted receptor tyrosine kinase inhibitors. J Med Chem 48:6066–6083
Bold G, Altmann KH, Frei J et al (2000) New anilinophthalazines as potent and orally well absorbed inhibitors of the VEGF receptor tyrosine kinases useful as antagonists of tumor-driven angiogenesis. J Med Chem 43:2310–2323
Zupan J, Novic M, Ruisánchez I (1997) Kohonen and counter propagation artificial neural networks in analytical chemistry. Chemom Int Lab Syst 38:1–23
Gasteiger J, Zupan J (1993) Neural networks in chemistry. Angew Chem Int Ed 32:503–527
Tropsha A, Gramatica P, Gombar VK (2003) The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb Sci 22:69–76
Lobanov V, Karelson M, Katriztky AR (1996) Codessa v.2.21 software package. Center for Heterocyclic Compounds, University of Florida, Gainesville
Ballabio D, Consonni V, Todeschini R (2009) The Kohonen and CP-ANN toolbox: a collection of MATLAB modules for self organizing maps and counterpropagation artificial neural networks. Chemom Intell Lab Syst 98:115–122
Schrödinger LLC (2005) MacroModel 91 reference manual. Schrödinger LLC, New York
Scherman W, Day T, Jacobson MP, Friesner RA, Farid R (2006) Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 49:534–553
Schrödinger LLC (2009) Glide, v.5.5. Schrödinger LLC, New York
Schrödinger LLC (2009) Prime, v.2.1. Schrödinger LLC, New York
Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA (2006) PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening. 1. Methodology and preliminary results. J Comput Aided Mol Des 20:647–671
Güner OF, Henry DR (1998) Formula for determining the “goodness of hit lists” in 3D database searches. Accelrys/MDL Information Systems, Inc., San Diego/San Leandro. http://www.netsci.org/Science/Cheminform/feature09.html
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 1721 kb)
Rights and permissions
About this article
Cite this article
Almerico, A.M., Tutone, M. & Lauria, A. Receptor-guided 3D-QSAR approach for the discovery of c-kit tyrosine kinase inhibitors. J Mol Model 18, 2885–2895 (2012). https://doi.org/10.1007/s00894-011-1304-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-011-1304-0