
Abstract
The strong collinear polarizability of the A–H bond in A–H⋅⋅⋅B hydrogen bonds is shown to lead to an enhanced σ-hole on the donor hydrogen atom and hence to stronger hydrogen bonding. This effect helps to explain the directionality of hydrogen bonds, the well known cooperative effect in hydrogen bonding, and the occurrence of blue-shifting. The latter results when significant additional electron density is shifted into the A–H bonding region by the polarization effect. The shift in the A–H stretching frequency is shown to depend essentially linearly on the calculated atomic charge on the donor hydrogen for all donors in which A belongs to the same row of the periodic table. A further result of the polarization effect, which is also expected for other σ-hole bonds, is that the strength of the non-covalent interaction depends strongly on external electric fields.

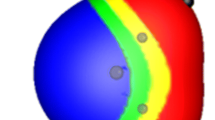
Contour diagram of the difference in MEP between water polarized by a point charge of magnitude −0.2762 at a distance of 1.946 Å and unperturbed water. The polarization is strongly directional along the O–HDonor bond vector
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The first mention of hydrogen bonds in the literature is generally attributed to Latimer and Rodebush in 1920 [1], although earlier work [2–5] mentions the phenomenon without naming it explicitly. The exact nature of hydrogen bonds, and more recently the relative importance of the “electrostatic” and “covalent” contributions, has been a subject of intense discussion ever since [6–10]. As with all such discussions, there are really no answers because the physically observable interaction cannot be partitioned uniquely into independent components whose very existence is due only to bonding theories.
Hydrogen bonding concepts continue to evolve [11–15], even to the extent that a new IUPAC definition has been proposed recently [16]. The old model, A–H⋅⋅⋅B, where A is an electronegative atom and B is a basic site, has now been extended to include C–H⋅⋅⋅B complexes and A–H⋅⋅⋅H–M “dihydrogen” bonds. A–H “blue-shifting” is accepted as a possibility; this is particularly significant because A–H “red-shifting” had previously been regarded as a universal characteristic of hydrogen bonds.
A satisfactory general theory of hydrogen bonding must be able to explain at least two prominent characteristics: its directionality and the dependence of the frequency of the donor A–H bond on the presence of a hydrogen bond. We have pointed out [17] that some of the directionality can be explained purely electrostatically because of the presence of a positive σ-hole [18, 19]. A σ-hole is a region of lesser electronic density on the side of an atom opposite to a covalent bond to that atom. If the electronic density is diminished sufficiently, a localized positive electrostatic potential results (a positive σ-hole), through which the atom can interact with a negative site [17–20]. Positive σ-holes have been found for covalently bonded hydrogens and Group IV–VII atoms. For Group VII, their interactions are called “halogen bonding.”
Recently, Truhlar et al. [21] discovered that most of the polarizability of organic molecules can be recovered in ab initio or density-functional theory (DFT) calculations by adding diffuse p-orbitals to hydrogen atoms. This is significant in the present context because the hydrogen σ-hole results from the polarization of the electron density formally belonging to the hydrogen atom away from the hydrogen nucleus into the A–H bond [17, 22, 23]. The basis for the results of Truhlar et al. [21] is therefore that the dominant polarization mechanism in compounds with A–H bonds is the translation of the “hydrogen” electronic density along the A–H bond vector, as shown schematically in Fig. 1.

Schematic diagram of the principal polarization mechanism in compounds containing bonds between non-hydrogen atoms (A) and hydrogen (H). Dashed line Position of the center of the electron density assignable to the hydrogen 1 s-orbital. The locations of the nuclei are indicated by A and H
This effect has long been recognized [22, 23]; it is the reason why standard crystallographic methods, which use buildups of electronic density to assign nuclear positions, typically underestimate hydrogen bond lengths by roughly 0.1 Å [24, 25]. However, we are not aware that its relevance for hydrogen bonding, or as the dominant polarization mechanism of molecules with A–H bonds, has been pointed out. However, Jug and Geutner [26] discovered that diffuse p-functions on the hydrogens are necessary to enable SINDO1 to describe hydrogen bonds, which may be an indication of the effect discussed here.
This polarization, which can be induced most effectively by a charge (e.g., a hydrogen-bond acceptor atom) collinear with the A–H bond, can have two effects; (1) to induce a more pronounced σ-hole, and (2) to shift electron density into the A–H bonding region. In the following, we investigate these two effects computationally, using simple point-charge models for hydrogen-bond acceptors, and draw attention to an interesting consequence. Hermannson [27, 28] has used a similar approach with both homogeneous electric fields and point charges to investigate the effect of hydrogen-bond acceptors on the electron density and vibrational frequencies of H-bond donors, and we have shown previously [17] that such models can reproduce the directionality of weak polar interactions.
Methods
All calculations were performed with the Gaussian’09 suite of programs [29], the aug-cc-pVTZ basis set [30, 31] and using a second order Møller-Plesset correction [32] for electron correlation with a restricted Hartree-Fock reference wavefunction (MP2/aug-cc-pVTZ). Geometries were fully optimized unless otherwise noted and normal vibrations were calculated within the harmonic approximation at the MP2/aug-cc-pVTZ level. Plots of electron densities and molecular electrostatic potentials (MEPs) used the MP2/aug-cc-pVTZ electron density. Point charges and electric fields were included in the calculations using the options available in Gaussian’09 [29].
Results and discussion
Polarization-induced σ-holes: water
Figure 2 shows how the MEP [33] of water, projected onto its 0.002 e Bohr−3 isodensity surface [34], depends on the magnitude of negative point charges placed at the position of the oxygen atom of an acceptor water molecule in the dimer [17].

Second order Møller-Plesset correction for electron correlation with a restricted Hartree-Fock reference wavefunction (MP2/aug-cc-pVTZ) molecular electrostatic potential (MEP) projected onto the standard isodensity surface (0.002 e Bohr−3) of water with point charges situated at the position of the H-bond acceptor oxygen in the water dimer. The magnitudes of the charges are (a) 0.0, (b) −0.2, (c) −0.4, (d) −0.6. The H-donor hydrogen atom is on the left and clearly shows the increasingly large σ-hole with increasing polarizing charge
The increasing size and strength of the σ-hole as the polarizing charge becomes more negative can be seen clearly (this σ-hole exists in unperturbed water but is weak [17]). This effect can be quantified by plotting the maximum (most positive) value of the MEP on the surface at the position of the σ-hole, V S,max against the magnitude of the polarizing charge, as shown in Fig. 3.

MEP on the surface at the position of the σ-hole, V S,max (kcal mol−1) plotted against the magnitude of the polarizing charge for the water dimer. For negative charges, V S,max is the maximum of the MEP on the entire surface; for positive charges, it is the maximum associated with the σ-hole on the donor hydrogen atom. Red point Charge corresponding to a second water molecule [17]
V S,max depends essentially linearly upon the magnitude of the inducing charge (and hence the field that it produces at the donor hydrogen). Most significantly, V S,max is 12.2 kcal mol−1 more positive (69.6 vs 57.4 kcal mol−1) for the calculation with the charge that corresponds [17] to a second water molecule than in the unperturbed molecule. The magnitude of this charge was determined by placing a point charge at the position of the acceptor oxygen in the water dimer and adjusting the size of the charge to obtain the calculated water dimerization energy [17]. As the strengths of hydrogen and halogen bonds have been shown to depend linearly on V S,max [35–39], this polarization should result in an approximately 20% increase in hydrogen-bond strength.
Because the polarization occurs along the A–H bond vector, it also induces directionality in the MEP around the donor hydrogen atom, as shown in Fig. 4. The polarization is limited largely to the O–HDonor bond and corresponds quite closely to a perturbation of the O–HDonor bond dipole. Some electron density is shifted to the distal hydrogen atom as a consequence of the general polarization from left to right in Fig. 5. The shift in electron density within the O–HDonor bond is most interesting and entirely consistent with the simple picture shown in Fig. 1. The induced σ-hole shows the expected [17, 18] directionality.

Contour diagram of the difference in MEP between water polarized by a point charge of magnitude −0.2762 at a distance of 1.946 Å and unperturbed water. The polarization is strongly directional along the O–HDonor bond vector

Contour diagram of the difference in the electron density (a.u.) between water polarized by a point charge of magnitude −0.2762 at a distance of 1.946 Å and unperturbed water
Polarization-induced σ-holes: CX3H
Figure 6 shows the effect of a negative point charge on the MEP at the isodensity surface of CF3H. Figure S1 in the electronic supplementary material shows the corresponding pictures for CCl3H. The induced area of positive MEP on the hydrogen donor collinear with the C–H bond is not as localized as that for water, suggesting that CF3H⋅⋅⋅A hydrogen bonds should be less strongly directional than their HOH⋅⋅⋅A equivalents. This is shown for complexes with a water molecule as H-bond acceptor in Fig. 7. The directional dependence for CF3H as H-bond donor is far weaker than for water, and the deepest minimum lies at a C-H⋅⋅⋅O angle of 124.7°.

MEP (kcal mol−1) at the standard isodensity surface of CF3H, demonstrating the effect of negative point charges on the electrostatic characteristics of the hydrogen atom. The magnitudes of the charges are (a) 0.0, (b) −0.2, (c) −0.4, (d) −0.6

Dependence of the energy of H-bonded complexes (relative to the minima) on the A–HDonor⋅⋅⋅O angle for the A–H⋅⋅⋅OH2 complexes (MP2/aug-cc-pVTZ Born-Oppenheimer energies)
The less delocalized nature of the CF3H σ-hole is not, however, the result of the polarization by the point charge, which is even more directional than that in water, as shown in Fig. 8. Once again, the characteristic polarization of the C–H bond is evident. This is indeed seen for the complex with a water molecule as the H-bond acceptor (Fig. 9). The directional dependence for CF3H as H-bond donor is far weaker than for water, and the deepest minimum lies at a C–H⋅⋅⋅O angle of 124.7°. This angle still allows the oxygen to interact with the expanded σ-hole, and also permits secondary stabilizing interactions between a water hydrogen and a fluorine, as shown in Fig. 9.

Contour diagram of the difference in the MEP (kcal mol−1) between CF3H polarized by a point charge of magnitude −0.2762 at a distance of 2.210 Å and an unperturbed CF3H molecule. The polarization is strongly directional along the C–H bond vector

MP2/aug-cc-pVTZ optimized structure of the CF3H complex with water showing the bent C–H–O linkage and the close O–H and H–F contacts
A–H stretching frequencies
Analogous point-charge models to those described above were used to investigate the effect of point charges at the position of an H-bond acceptor on the O–H stretching frequency in methanol. The results are shown in Fig. 10.

Dependence of the harmonic O–H stretching frequency in methanol on the magnitude of the perturbing point charge at the position of a water H-bond acceptor. The horizontal reference line shows the frequency in CH3OH⋅⋅⋅OH2
The effect of the charge on the frequency is approximately parabolic, as also found by Hermansson [27, 28]. Point charges between 0 and +0.5 can induce a blue shift in the O–H bond stretching frequency, but this is not relevant to real hydrogen-bonding situations. The calculated O–H bonding shift from CH3OH⋅⋅⋅OH2 is reached for a point charge of −0.55, approximately double that required to mimic an acceptor water molecule energetically [17]. This suggests that a purely electrostatic model cannot reproduce the entire red shift found for “normal” hydrogen bonds. This is consistent with Hermansson’s conclusions [27, 28].
The C–H stretching frequencies in CF3H and CCl3H behave differently, as shown in Fig. 11. The C–H stretching frequencies show the opposite trend to that found for O–H (Fig. 10). Negative point charges induce a blue shift, as also found by Hermansson [27, 28]. Interestingly, the flat maxima found for the C–H stretching frequencies, at charges around −0.4 to −0.5, correspond closely to the calculated C–H frequencies in the linear CX3H⋅⋅⋅OH2 complexes, and the charges on the oxygens in the complexes are very similar to the point charges that give the maxima in Fig. 11. It is a consistent feature of the O–H and C–H stretching frequencies that they attain the same value with a point charge of −0.5 as in the complex with water (Figs. 10, 11).

Dependence of the harmonic C–H stretching frequencies in trifluoro- and trichloromethane on the magnitude of the perturbing point charge at the position of the O atom in the linear CX3H⋅⋅⋅OH2 complex. The horizontal reference lines show the calculated frequencies in linear CX3H⋅⋅⋅OH2
Field-dependence of hydrogen bonding
The above analysis suggests that, because the polarization of the A–HDonor bond affects the hydrogen bonding strength, directionality and the A–HDonor stretching frequency, all three properties should be particularly sensitive to external fields applied collinear to the A–HDonor bond. This can be tested computationally. Figure 12 shows the results. As expected, the hydrogen-bond energy depends strongly (and approximately linearly) on the applied field. Shifting the “hydrogen” electron density into the O–H bond increases V S,max and therefore strengthens the hydrogen bond. As V S,max depends essentially linearly on the applied charge (Fig. 3), this behavior is analogous to that found previously for halogen bonding [35–37].

Effect of an external electric field on the hydrogen-bond strength in the water dimer. For details of the calculations, see the electronic supplementary material. The field is oriented along the donor O–H bond with the positive direction running from O (positive) to H (negative)
As expected from the discussion above, external fields that increase the polarization of the O–H donor bond (i.e., shift the hydrogen electron density towards O) increase the strength of the hydrogen bond, and those in the reverse direction weaken it. To put the magnitude of the applied fields into perspective, the field at the nucleus of the donor hydrogen atom is very close to 0.01 a.u. at MP2/aug-cc-pVTZ, so that the external field range shown in Fig. 11 extends from essentially cancelling the inherent field at this position to doubling it. The plot is almost linear with a slope of −147 kcal mol−1 a.u.−1.
Discussion
Truhlar et al.’s discovery that polarization of A–H bonds accounts for most of the polarizability of organic molecules [21] opens a new perspective on hydrogen bonding. Shifting the electron density assignable to the donor hydrogen atom towards the non-hydrogen atom A leads to an area of positive electrostatic potential that is collinear and opposite to the A–H bond. This positive cap resembles the σ-hole found for halogens and atoms of Groups IV–VI [17–19, 40–43], although the rationalization for their occurrence is different because our previous arguments relied on hybridization, which formally does not occur for hydrogen. This hydrogen σ-hole strengthens hydrogen bonding, but also increases the directionality of hydrogen bonds as it becomes stronger. Thus, the simple picture illustrated in Fig. 1 can help to explain many important features of hydrogen bonding.
We have shown that the σ-hole of a donor hydrogen can be made more positive by the polarizing effect of a negative point charge. This polarization-induced strengthening of σ-holes should also occur for covalently bonded halogens and atoms of Groups IV–VI. This can explain some otherwise puzzling observations. For instance, the chlorine in H3C–Cl has a σ-hole potential of near zero, perhaps slightly negative [19]. Yet H3C–Cl has been found computationally to form a complex with formaldehyde, H3C–Cl⋅⋅⋅O = CH2, with an interaction energy of about −1.2 kcal mol−1 [44]. It was suggested earlier [19] that this may be a case of the base, O = CH2, inducing a significantly positive σ-hole on the chlorine. Our present results support this interpretation.
The origin of the blue shift in “improper” hydrogen bonds [11] has been discussed very thoroughly in the literature. Hermansson [27, 28] has also examined the dependence of the A–H stretching frequency (in her case anharmonic) on an external electric field, both uniform and arising from an external point charge with similar results to those described above. In this work, the origin of the blue shift was analyzed very carefully and three conditions for its observation were defined:
-
1.
A negative dipole-moment derivative, \( \frac{{d\mu (0)}}{{d{r_{{A - H}}}}} \), for the A–H bond length (μ (0) is the permanent dipole moment of the A–H molecule, in the absence of a field),
-
2.
An interaction with any electron-density concentration on an H-bond acceptor and
-
3.
An additional blue shift due to electronic exchange overlap.
Condition 1 is consistent with our observations, but neither 2 nor 3 apply in our case as the point charges used in this work to represent the H-bond acceptor have no electrons. However, we noted above that charges approximately twice as large as those needed to reproduce the hydrogen-bond energy [17] are needed to reproduce the observed frequency shifts. We can therefore conclude that purely electrostatic effects can reproduce the observed directionality and frequency shifts of hydrogen bonds, but that, as concluded by Hermansson, electronic exchange overlap probably also contributes to the frequency shift in the same direction as the electrostatic effect (see discussion above).
Earlier, we investigated blue vs red shifts in σ-hole bonded complexes A–Y⋅⋅⋅B, where Y is an atom of Groups V–VII [45], in terms of Hermansson’s formula [27] for the A–Y frequency change due to a uniform electric field. This allowed us to explain the increase or decrease in the A–Y frequency that was obtained computationally for each complex, B being NH3 and/or HCN. We found, as above, that one requirement for a blue shift is that the derivative of the σ-hole molecule’s permanent dipole moment be opposite in direction to the electric field due to the base B. A second requirement that also has to be satisfied is that the induced dipole moment derivative be less than half of the permanent dipole derivative. This means that the field created by the base should not be too strong [27]. Thus, O2N–Cl had a blue shift in O2N–Cl⋅⋅⋅NCH but a red shift in O2N–Cl⋅⋅⋅NH3, because NH3 produces a stronger electric field than does HCN [45]. (A subsequent application of Hermansson’s formula was to predict the effects of external electric fields upon “trigger linkage” bonds in some prototypical energetic molecules [46]. It was found that fields that reinforce the intrinsic polarity strengthen the trigger linkages, and fields in the reverse direction weaken it; the same conclusion was reached in the present work for hydrogen bonds.)
The derivative of the bond dipole moment is neither an intuitive nor a common quantity in qualitative bonding theory, so we have tried to relate it to more familiar concepts. Figure 13 shows that the calculated frequency shift exhibits a simple dependence on the natural bond orbital (NBO) charge [47] on the donor hydrogen atom calculated at the MP2/aug-cc-pVTZ level.

Plot of the calculated shift in the frequency of the A–H bond stretch on forming a hydrogen-bonded complex with an acceptor water molecule against the natural bond orbital (NBO) charge on the donor hydrogen (both at the MP2/aug-cc-pVTZ level). The lines are the best least-squares fit for all molecules in which A belongs to either the first or the second row of the periodic table
(CH3)2PH does not act as a hydrogen-bond donor in its complex with water, but rather as a σ-hole donor at phosphorus, and so the P–H⋅⋅⋅O angle was constrained to be linear for the data shown in Fig. 13. For molecules in which A belongs to the same group in the periodic table, there is a surprisingly good relationship between the NBO-charge on the donor hydrogen and the shift in the A–H stretching frequency on forming a hydrogen bond with water. Because of the limited range available for compounds in which A is a first-row element, the relationship between the charge on the donor hydrogen and the frequency shift appears to be linear, but clearly the shift must approach zero as the hydrogen bond becomes weaker, so that the polynomial fit shown for the second-row elements is more appropriate.
These results suggest that Hermansson’s first criterion can also be expressed in terms of the net atomic charge on the donor hydrogen atom. To a first approximation, we can expect an A–H blue shift for H-bond donors in which the donor hydrogen has an NBO charge less positive than approximately +0.2 e at MP2/aug-cc-pVTZ for H-bond donors in which A is a first-row element. The corresponding maximum positive charge on the donor hydrogen for second-row donors is approximately +0.06 e, making blue-shifted hydrogen bonds far less likely for second-row donors.
The observed field effects on the strengths of hydrogen bonds can be rationalized as above by the shift of the center of the “hydrogen” electron density along the A–H bond with its concomitant effect on the magnitude of the σ-hole potential on the donor hydrogen, as shown in Fig. 1. This view is pictorially attractive but equivalent to one in which the effect of the field is to polarize the A–H bond dipole, resulting in a stronger dipole-acceptor interaction. The novel feature of the present interpretation for hydrogen bonds is that the work of Truhlar et al. [21] suggests that the major part of the polarization of molecules with A–H bonds can be represented as A–H-bond polarization along the direction of the bonds. Thus, the greatest induced dipoles for a correctly oriented field are those that also most affect the strengths of hydrogen bonds. This is, of course, the origin of the well-known cooperative effect in hydrogen bonding. [48, 49] As, however, strong electric fields are found in host cavities such as those of zeolites [50], enzymes or biological receptors [51], we can expect that the strengths of hydrogen bonds, usually assumed to be transferable and additive in, for instance, biological scoring functions, are actually quite strongly dependent on the local field in the host. Interestingly, this effect is also consistent with the known [52] dependence of the chemical potential on the electrostatic potential and field at the nucleus (in this case of the donor hydrogen).
Finally, we are aware that there are any number of other ways to interpret the computational experiments that we describe above. This will always be the case with qualitative descriptions of bonding interactions. We have limited our discussion to simple polarization arguments and electrostatic interactions in order to provide as simple a picture as possible. Even this approach leads to the important prediction that hydrogen bonds in the cavities of polar hosts are affected strongly by the external electric field of the host. We are now testing this hypothesis for binding sites in proteins.
References
Latimer WM, Rodebush WH (1920) J Am Chem Soc 42:1419–1433
Werner A (1902) Liebigs Ann Chem 322:261–296
Werner A (1903) Chem Ber 36:147–159
Moore TS, Winmill TF (1912) J Chem Soc 101:1635–1676
Pfeiffer P, Fischer P, Kunter J, Monti P, Pros Z (1913) Liebigs Ann Chem 398:137–196
Arunan E (1999) Curr Sci 77:1233–1235
Scheiner S (1997) Hydrogen bonding. Oxford University Press, Oxford
Jeffrey GA (1997) Introduction to hydrogen bonding. Oxford University Press, Oxford
Desiraju G, Steiner T (1997) The weak hydrogen bond. Oxford University Press, Oxford
Muller-Dethlefs K, Hobza P (2000) Chem Rev 100:143–168
Hobza P, Havlas Z (2000) Chem Rev 100:4253–4264
Belkova NV, Shubina ES, Epstein LM (2005) Acc Chem Res 38:624–631
Grabowski SJ (ed) (2006) Hydrogen bonding—new insights. Springer, Dordrecht, The Netherlands
Grabowski SJ, Sokalski WA, Leszczynski J (2006) Chem Phys Lett 432:33–39
de Oliveira BG, Ramos MN (2010) Int J Quantum Chem 110:307–316
IUPAC Reports (2011) http://media.iupac.org/reports/provisional/abstract11/arunan3_0311.html, accessed 5 May 2011
Murray JS, Riley KE, Politzer P, Clark T (2010) Aust J Chem 63:1598–1607
Clark T, Hennemann M, Murray JS, Politzer P (2007) J Mol Model 13:291–296
Politzer P, Murray JS, Clark T (2010) Phys Chem Chem Phys 12:7748–7757
Murray JS, Lane P, Clark T, Riley KE, Politzer P (2011) J Mol Model. doi:10.1007/s00894-011-1089-1
Fielder L, Gao J, Truhlar DG (2011) J Chem Theor Comput 7:852–856
Ermer O, Lifson S (1973) J Am Chem Soc 95:4121–4132
Burkert U, Allinger NL (1972) Molecular mechanics—ACS Monograph 177. American Chemical Society, Washington DC
Wilson CC (2000) Single crystal neutron diffraction from molecular materials. World Scientific, Singapore
Parkin A, Harte SM, Goeta AE, Wilson CC (2004) New J Chem 28:718–721
Jug K, Geutner G (1993) J Comput Chem 14:639–646
Hermansson K (2002) J Phys Chem A 106:4695–4702
Pejov L, Hermanssno K (2003) J Chem Phys 119:313–324
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision A.02. Gaussian Inc, Wallingford CT
Dunning TH Jr (1980) J Chem Phys 90:1007–1023
Kendall RA, Dunning TH Jr, Harrison RJ (1992) J Chem Phys 96:6796–6806
Head-Gordon M, Pople JA, Frisch MJ (1988) Chem Phys Lett 153:503–506
Politzer P, Truhlar DG (eds) Chemical applications of atomic and molecular electrostatic potentials. Plenum, New York
Bader RFW, Carroll MT, Cheeseman JR, Chang C (1987) J Am Chem Soc 109:7968–7679
Riley KE, Murray JS, Concha MC, Hobza P, Politzer P (2009) J Chem Theor Comput 5:155–163
Shields ZP, Murray JS, Politzer P (2010) Int J Quantum Chem 110:2823–2832
Riley KE, Murray JS, Fanfrlík J, Řezáč J, Solá RJ, Concha MC, Ramos FM, Politzer P (2011) J Mol Model. doi:10.1007/s00894-011-1015-6
Murray JS, Politzer P (1991) J Org Chem 56:6715–6717
Hagelin H, Murray JS, Brinck T, Berthelot M, Politzer P (1995) Can J Chem 73:483–488
Murray JS, Lane P, Clark T, Politzer P (2007) J Mol Model 13:1033–1038
Murray JS, Lane P, Politzer P (2007) J Quantum Chem 107:2286–2292
Murray JS, Lane P, Politzer P (2009) J Mol Model 15:723–729
Politzer P, Murray JS (2009) An overview of σ-hole bonding: an important and widely occurring noncovalent interaction. In: Leszczynski J, Shukla MK (eds) Practical aspects of computational chemistry. Springer, Amsterdam
Riley KE, Hobza P (2008) J Chem Theor Comput 4:232–242
Murray JS, Concha MC, Lane P, Hobza P, Politzer P (2008) J Mol Model 14:699–704
Politzer P, Murray JS, Lane P (2009) Int J Quantum Chem 109:534–539
Reed AE, Weinstock RB, Weinhold F (1985) J Chem Phys 83:735–746
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926
See, for instance Parker LL, Houk AR, Jensen JH (2006) J Am Chem Soc 128:9863–9872
Barrachin B, Cohen de Lara E (1986) J Chem Soc. Faraday Trans 2(82):1953–1966
Nakagawa K, Suzuki S, Fujii R, Gardiner AT, Cogdell RJ, Nango M, Hashimoto H (2008) J Phys Chem B 112:9467–9475
Politzer P, Clark T (2005) Mol Phys 10:891–895
Acknowledgments
We thank the Deutsche Forschungsgemeinschaft for funding as part of SFB583 “Redox-Active Metal Complexes” and the Excellence Cluster “Engineering of Advanced Materials”.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Supporting material available
The full text of reference 29, MEP surfaces for CCl3H and details (Gaussian archive entries) of the calculations are available free of charge via at http://pubs.acs.org.
Esm 1
(DOC 49 kb)
Rights and permissions
About this article
Cite this article
Hennemann, M., Murray, J.S., Politzer, P. et al. Polarization-induced σ-holes and hydrogen bonding. J Mol Model 18, 2461–2469 (2012). https://doi.org/10.1007/s00894-011-1263-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-011-1263-5