
Abstract
First-principles calculations were performed for fluorine-decorated graphene (fluorographene). Three different hexagonal clusters were used—circular (C24H12), triangular (C23H10) and rectangular (C24H12)—and the fluorine atoms were randomly distributed in the mesh. Graphene is structurally stable in the three geometries, but fluorographene stability is only attained for the circular and triangular clusters. Gaps of the circular graphene and the corresponding fluorographene are 2.94 and 1.13 eV, respectively; in the triangular case, the values are zero and 0.47 eV. Both the circular and triangular structures show a transition from ionic to covalent character.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Six years ago, Geim and coworkers [1] performed the first research on two-dimensional nanostructures. These systems are attractive due their large number of applications, and efforts have been made to improve their performance. Recently, decorating the graphene surface gave rise to new structures, including graphane [2, 3], graphone [4], graphene oxide [5, 6], graphanol [7], and the recently reported fluorographene [8]. Fluorographene is graphene doped with fluorine atoms and has an experimental band gap of 3.0 eV and a Young’s modulus of 100 N/m. Of course, information on this system is scarce, and it would be interesting to explore its potential applications. This means that basic research is necessary in order to determine its main properties. For example, it would be interesting to know the influence of the geometry on parameters like the dipole moment, vibrational frequencies, binding energy, and the difference between the HOMO and the LUMO (band gap). Therefore, in this work, using first-principles calculations, we investigate the electronic properties of the graphene when is decorated with fluorine atoms (analogous to fluorocarbons). We study the effect of randomly decorating the graphene with F atoms on the geometry.
Computational procedure
In order to study the fluorine-decorated graphene and the influence of the geometry on its properties, we employed three different clusters, namely a circular armchair cluster (C24H12; Fig. 1a), a triangular zigzag cluster (C23H10; Fig. 1b), and a rectangular armchair cluster (C24H12; Fig. 1c). For the circular and the triangular sheets, we considered the addition of one F atom to the mesh; for the rectangular sheet, we studied the cases where one, two, three, four, and five F atoms were added. The circular cluster model has been successfully used by Chigo Anota et al. [9–14; Chigo Anota et al. (2010) On the electronic properties of the two dimensional honeycomb GaInN and GaAlN alloys, submitted to Eur Phys J D] to study various two-dimensional systems. Calculations were performed using density functional theory (DFT) [15–18], as implemented in the DMOL3 code available from Accelrys Inc. [19]. We utilized the LDA approximation for the exchange-correlation term within the parameterization of Perdew–Wang (PWC) [20]. The limit for the orbital was 0.40 nm; the convergence for the SCF cycles was 1.0 × 10−6 Ha. The realization of non-negative frequencies was the criterion for structural stability [21]. Additional details on the calculations can be found in [9–14; Chigo Anota et al. (2010) On the electronic properties of the two dimensional honeycomb GaInN and GaAlN alloys, submitted to Eur Phys J D].

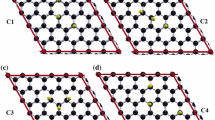
Initial geometry of graphene decorated with fluorine atoms: a circular structure, b triangular structure, c rectangular structure
Results and discussion
First, we optimized the graphene in each of the three models. All of them exhibited positive vibration frequencies and thus were structurally stable; the C–C bond lengths in the three graphene geometries were all 1.41 Å. Figure 1 shows the initial (a–c) and the optimized (e–g) structures when graphene is decorated with four fluorine atoms. Only the circular and the triangular geometries are stable, forming σ and π bonds; the C–C bond length increases to 1.44 Å for the circular cluster and remains the same as for graphene for the rectangular one. The C–F bond length is very similar for all stable geometries (1.43 Å), and the C–H bond length is 1.09 in all cases. When the number of fluorine atoms is 12, the circular structure becomes unstable. The C–C bond length is very similar to those of graphane [2, 3], graphone [4], and graphanol [7]; see Table 1. The initial geometries are planar but the optimized structures are curved: the circular sheet has an angle of curvature of 27.91° (Fig. 1e), the curvature of the triangular sheet is 32.61° (Fig. 1f), and the curvature of the rectangular (unstable) sheet is 34.37° (Fig. 1g). We can see that although the rectangular sheet is unstable, it presents the highest curvature. We then decorated this sheet with one, two, three, and five F atoms and calculated the stabilities of the resulting structures and their angles of curvature. Figure 2 shows the initial and the optimized structures for these cases; all of them are stable, and the angle of curvature increases as the number of F atoms increases, reaching a value of 59.76° for five F atoms. All this may suggest a methodology for growing tubular structures starting from graphene.

Initial (a, c, e, g) and final (b, d, f, h) geometries of graphene decorated with five, three, two and one fluorine atoms, respectively
From Table 1, we can see that graphene has almost no dipole moment in all the geometries (i.e., all of them are ionic). When these structures are doped with F atoms, the dipole moment increases, indicating that the system has covalent character. In the case of the rectangular sheet, there is a systematic increase in this parameter when the number of F atoms is increased from one to five; only one F atom is needed to significantly modify the polarity of graphene. The binding energy has almost the same value for all three clusters of graphene, and a high value when the F atoms are added to the mesh; in the case of the rectangular cluster, the binding energy increases as the number of F atoms increases. According to our results, it is not possible to obtain a rectangular graphene decorated with four F atoms, but it is possible to grow rectangular graphene decorated with one, two, three and five F atoms.
The circular and rectangular graphene arrays show a gap between the HOMO and the LUMO, in contrast to the triangular one, which has no gap. When the F atoms are added, the gap is reduced in the circular case and the corresponding value for the triangular case increases a little (0.47 eV); the experimental value for fluorographene is 3.0 eV, indicating that it shows semiconductor behavior, in agreement with the results obtained in this work. Something similar happens when graphene is decorated with hydrogen: graphane (3.5 eV), graphone (0.46 eV), and graphanol (3.22 eV). Other systems, like BN, present different behavior when they are decorated with hydrogen; BN changes from being a semiconductor to a semimetal. The gap of rectangular graphene without any F atoms is 0.75 eV. When one F atom is added to it, this parameter decreases to 0.06 eV; i.e., the sheet changes from being a semiconductor to a metal, and only one F atom is needed to change the character of the sheet. As two, three, and four F atoms are added, the gap increases monotonically, but when five F atoms have been inserted it begins to decrease.
Conclusions
We have analyzed the influence of geometry on the electronic properties of F-decorated clusters of graphene (fluorographene). In this study, randomly distributed F atoms were considered: four for circular and triangular clusters; one, two, three, four, and five for a rectangular cluster. The circular and triangular fluorographene geometries are stable, and the lengths of the C–C and C–H bonds do not change significantly upon the addition of F atoms. Both the circular and triangular systems have almost the same C–F bond length (1.43 Å). A transition from ionic (graphene) to covalent (F-decorated graphene) behavior is observed. In the case of the circular cluster, the gap between the HOMO and the LUMO decreases from 2.94 to 1.13 eV; for the triangular cluster, this quantity increases from zero to 0.47 eV. Rectangular fluorographenes with one, two, three, four and five F atoms exhibit sheet curvature; this curvature, the dipole moment of the fluorographene, and the binding energy all increase as the number of F atoms increases. Changes in the band gap are less than 1 eV. All of these properties of fluorographene make it a candidate for use in optoelectronic applications, and may suggest a possible methodology for obtaining nanotubes by adding F atoms to the graphene.
References
Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA (2004) Science 306:666–669
Sofo JO, Chaudhari AS, Barber GD (2007) Phys Rev B 75:153401–153404
Elias DC, Nair RR, Mohiuddin TMG, Morozov SV, Blake P, Halsall MP, Ferrari AC, Boukhvalov DW, Katsnelson MI, Geim AK, Novoselov KS (2006) Science 323:610–613
Zhou J, Wang Q, Sun Q, Chen XS, Kawazoe Y, Jena P (2009) Nano Lett 9:3867–3870
Dikin DA, Stankovich S, Zimney EJ, Piner RD, Dommett GHB, Evmenenko G, Nguyen ST, Ruoff RS (2007) Nature 448:457–460
Hernández Rosas JJ, Ramirez Gutierrez RE, Escobedo Morales A, Chigo Anota E (2010) J Mol Model. doi:10.1007/s00894-010-0818-1
Wang WL, Kaxiras E (2010) On graphene hydrate. http://arxiv.org/pdf/1003.3086
Nair RR, Ren WC, Jalil R, Diaz I, Kravets VG, Britnell L, Blake P, Schedin F, Mayorov AS, Yuan S, Katsnelson MI,Cheng HM, Strupinski W, Bulsheva LG, Okotrub AV, Novoselov KS, Geim AK, Grigoreva IV, Grigorenko AN (2010) Fluorographene: mechanically strong and thermally stable two-dimensional wide-gap semiconductor. http://arxiv.org/pdf/1006.3016v1
Chigo Anota E, Salazar Villanueva M (2009) Sup y Vac 22:23–28
Chigo Anota E, Hernández Cocoletzi H, Bautista Hernández A, Sanchéz Ramírez JF (2010) J Comput Theor Nanosci (accepted)
Chigo Anota E, Salazar Villanueva M, Hernández Cocoletzi H (2010) Phys Stat Solidi C 7:2252–2254
Chigo Anota E, Salazar Villanueva M, Hernández Cocoletzi H (2010) Phys Stat Solidi C 7:2559–2561
Chigo Anota E, Salazar Villanueva M, Hernández Cocoletzi J (2011) J Nanosci Nanotechnol. doi:10.1166/jnn.2011.3441
Chigo Anota E, Hernández Cocoletzi H, Rubio Rosas E (2010) Eur Phys J D (accepted)
Kohn W, Becke AD, Parr RG (1996) J Phys Chem 100:12974–12980
Jones RO, Gunnarsson O (1989) Rev Modern Phys 61:689–746
Kohn W (1999) Rev Modern Phys 71:1253–1266
Chigo Anota E, Rivas Silva JF (2005) Rev Col Fís 37:405–417
Delley B (1990) J Chem Phys 92:508–517
Perdew JP, Wang Y (1992) Phys Rev B 45:13244–13249
Foresman JB, Frisch Æ (1996) Exploring chemistry with electronic structure methods, 2nd edn. Gaussian Inc., Pittsburgh, p 300
Wang Y (2010) Phys Stat Solidi (RRL) 4:34–36
Acknowledgments
This work was partially supported by Vicerrectoria de Investigación y Estudios de Posgrado-Benemérita Universidad Autónoma de Puebla (CHAE-ING10-I), Facultad de Ingeniería Química- Benemérita Universidad Autónoma de Puebla (2009-2010), Cuerpo Académico Ingeniería en Materiales (BUAP-CA-177) and Consejo Nacional de Ciencia y Tecnología, México (grant no. 0083982).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Contreras, C.N., Cocoletzi, H.H. & Anota, E.C. Theoretical study of the electronic properties of fluorographene. J Mol Model 17, 2093–2097 (2011). https://doi.org/10.1007/s00894-010-0914-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-010-0914-2