
Abstract
The single mutations D30N and I50V are considered as the key residue mutations of the HIV-1 protease drug resistance to inhibitors in clinical use. In this work, molecular dynamics (MD) simulations combined with the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method have been performed to investigate the drug-resistant mechanisms of D30N and I50V to an inhibitor TMC-114. The analyses of absolute binding free energies using the separate trajectory approach suggests that the decrease in the van der Waals energy and electrostatic energy in the gas phase results in the drug resistance of D30N to TMC-114, while for I50V, the decrease in the electrostatic energy mainly drive its drug resistance to TMC-114. Detailed binding free energies between TMC-114 and individual protein residues are computed by using a per-residue basis decomposition method, which provides insights into the inhibitor-protein binding mechanism and also explains the drug-resistant mechanisms of mutations D30N and I50V to TMC-114. The study shows that the loss of the hydrogen bond between TMC-114 and the side chain of Asn30′ is the main driving force of the resistance of D30N to TMC-114, and in the case of I50V, the increase in the polar solvation energies between TMC-114 and two residues Val50′ and Asp30′ definitively drives the resistance of I50V to TMC-114. We expect that this work can provide some helpful insights into the nature of mutational effect and aid the future design of better inhibitors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The human immunodeficiency virus type І aspartic protease (PR) plays an essential role in the life cycle of the HIV virus. The function of PR is to cleave viral polyproteins into mature and functional HIV viral particles that can infect a host cell [1]. The PR is a C2-symmetric homo-dimer that consists of two identical 99 amino acid monomers. The active site, with the catalytic aspartic acids Asp25 and Asp25′, is located at the interface between the two monomers. The flaps of the PR are positioned over the active site and undergo major structural changes on binding of either a substrate or an inhibitor. HIV-1 protease inhibitors (PIs) that bind to the PR can inhibit the activation of the protease and hold back the replications of HIV virus [2]. Therefore, the PR has been an attractive target for antiretroviral therapy over the years.
So far, the Food and Drug Administration (FDA) has approved several antiviral inhibitors such as saquinavir [3], ritonavir [4], indinavir [5], nelfinavir [6], amprenavir [7], lopinavir [8], and atazanavir [9], which are used for the treatment of HIV infections. Several other PIs are in development. Despite the initial success of these FDA approved PIs, unfortunately, increasing viral resistance has reduced the binding affinities between inhibitors and the protease, and combined with unfavorable side effects, the therapeutic success and efficacy of the current inhibitors are highly limited [10–12]. Therefore, how to solve these problems have been great challenges in treating HIV.
Currently, drug-resistant mutations have been the most disturbing problems in the treatment of HIV infections [13, 14]. Resistance to the PIs is the consequence of amino acid substitutions that emerge either inside the substrate-binding pocket or at distant sites. If a substitution changes the shape or size of the binding cavity, the PI significantly loses its inhibiting potency. On the molecular level, drug resistance is primarily produced by a sharp decrease in the affinity of the PI for PR mutants, while the binding of the substrates is not heavily influenced. HIV patients in the world are looking forward to new potent PIs, which must be provided with low susceptibility to resistant mutations and fewer side effects than the existing drugs [15].
TMC-114, a next-generation nonpeptidic PI, is extremely potent against a wide spectrum of HIV strains in vitro, which include a variety of PI-resistant clinical strains. The stereochemistry of the terminal bis-tetrahydrofuran (bis-THF) moiety in TMC-114 has shown more polar interactions with main chain atoms of the PR, because two bis-THF oxygen atoms form hydrogen bonds with the backbone N-H functions of residues Asp30 (Asp30′) [16]. The structure of TMC-114 is shown by Fig. 1. Recently, several studies concerning the drug resistance of PR mutants on TMC-114 have been carried out. Irene T Weber et al. used the crystallographic method to analyze effectiveness of TMC-114 on the PR with highly drug-resistant mutations D30N, I50V, and L90M [17], they concluded that the D30N and I50V mutants resulted in the drug resistance to TMC-114, while the L90M mutant produced a tighter binding of TMC-114 to the PR. Kanda Nivesanond and co-workers used the Monte Carlo search method and quantum mechanical calculations to perform the conformational study of TMC-114, they discovered that the conformation of TMC-114 in the PR with a triple mutant (L63P/V82T/I84V) are close to the conformation observed in the X-ray complexes of TMC-114 and the wild-type PR [18].

Molecular structures of an inhibitor TMC-114

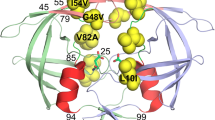
The structure of the PR/TMC-114 complex: The PR is shown in black ribbons. The sites of mutation are indicated using a ball-and-stick representation for Asp30 and Ile50. Catalytic aspartate residues are also shown in the ball-and-stick representation. Only one subunit is labeled
In order to investigate the molecular mechanism of drug resistance and evaluate the potency of TMC-114 to combat resistance, we choose two mutations for drug-resistant analysis (Fig. 2), D30N and I50V, which have generated a high level of resistance in clinical treatments [17]. D30N was selected for study since it is located nearby the active site and a major mutation exclusively selected for resistance to nelfinavir [19]; the mutation I50V was chosen because it is located in the flap region of the PR, and is the primary mutation that appears during the treatment with amprenavir, which is similar to TMC-114 structurally. In fact, it is important for the design of high potent inhibitors toward drug-resistant protease mutants to understand the mechanism of drug resistance at the atomic level.
In this study, nanosecond-level molecular dynamics (MD) simulations combined with free energy calculations have been carried out to obtain information about the binding of TMC-114 to the wild-type PR and the muted PR. Absolute binding free energy calculations were performed using the MM-PBSA method [20–28]. Detailed binding free energies between TMC-114 and individual PR residues are calculated by using a per-residue basis decomposition method [29]. The information of detailed interaction energies combined with the analysis of hydrogen bonds through the MD simulations provides insights into the PI-PR binding mechanisms and also help elucidate the drug-resistant mechanism of mutations to TMC-114 with the interpretation of the structural and energetic results from the simulation.
Materials and methods
The crystal structures of the wild-type and mutant PR complexed with TMC-114 were obtained from the Protein Date Bank (PDB). The PDB entries are: 1T3R for the wild-type PR (WT), 2F80 for the PR with D30N (PRD30N) and 2F8G for the PR with I50V (PR50V). There are alternate conformations in 2F80 and 2F8G: conformation A and B, owing to C2 symmetry of PR, only conformation B was selected for the starting model. Due to the importance of the protonation of Asp25/Asp25′ in the PR, only the mono-protonated PR was considered [30] and a proton was added to the oxygen atom OD1 of Asp25 [31, 32], which is the closest oxygen to TMC-114. All missing hydrogen atoms of the protein were added using the tleap module in the AMBER 9.0 software package [33]. All crystal water molecules in the crystal structure of the PI/PR complex were kept in the starting model. The force field ff03 was applied to produce the force field parameters of the protein and crystal water molecules. The electrostatic potential of TMC-114 was calculated by using the Gaussian 98 package [34] at the HF/6-31G* level. Atom-centered partial charges were derived by using the RESP [35] fitting technique in the AMBER. The general AMBER force field (GAFF) [36] was used to obtain the force field parameters of TMC-114, including the Lennard-Jones, torsion, bond angle terms.
A total of nine systems, including three PR/TMC-114 complexes, three free proteases and three inhibitors, were simulated in this work. To neutralize the charge of systems, an appropriate number of chloride counterions were placed to grids with the largest positive Coulombic potentials around the complexes and the proteases, and then the whole system was soaked in a truncated octahedral periodic box of TIP3P water molecules [37]. The distance between the edges of the water box and the closest atom of the solutes was at least 10 Å.
During the minimization and MD simulations, particle mesh Ewald (PMD) [38] was employed to treat the long-range electrostatic interactions in a periodic boundary condition. The Langevin dynamics with a collision frequency of 1.0 ps−1 was adopted to control the temperature of the systems. The SHAKE method [39] was used to constrain hydrogen atoms. The time step for all MD simulations is 2 fs, with a direct-space, non-bonded cutoff of 10 Å. Initial velocities were assigned from a Maxwellian distribution at the initial temperature. Before MD simulations, the entire system was subject to energy minimization in two stages to remove bad contacts between the complex and the solvents molecules. Firstly, the water molecules were minimized by holding the solute fixed with a harmonic constraint of a strength of 100 kcal mol−1 Å−2. Secondly, the entire system was minimized without restriction, and each stage was performed using the steepest descent minimization of 500 steps followed by a conjugate gradient minimization of 2500 steps. The system was then heated from 0 to 300 K in 100 ps and equilibrated at 300 K for another 100 ps. After the minimization and heating, 3-ns dynamics simulations without restriction were performed at a constant temperature of 300 K and a constant pressure of 1 atm. Finally, root-mean-square deviation (RMSD) of the protease Cα atom was computed from the MD trajectory relative to the initial structures to judge the stabilization of the system.
For each complex, a total number of 100 snapshots were taken from the last 800 ps on the MD trajectory with an interval 8 ps. The MM-PB/SA method and nmod module, which is implemented in Amber9 [33], were applied to compute the binding free energy of inhibitor to the protease. In this approach, the binding free energies (ΔG) are approximated by:
where ΔEMM is the molecular mechanics free energy in gas phase, ΔGsol is the solvation free energy, and TΔS is a term involving the entropy effect. The molecular mechanics free energy (ΔGMM) can further be expressed as:
where ΔEvdw and ΔEele represent the van der Waals and electrostatic interactions in gas phase, respectively. The solvation free energy (ΔGsol) is further divided into two components:
where ΔGpb and ΔGnp are the polar and non-polar contributions to the solvation free energy, respectively. The former component was computed using the pbsa program. The dielectric constant inside the solute was set to 1.0 and 80.0 in the solvent in our calculations. Whereas the latter term was determined by:
where SASA is the solvent-accessible surface area and was calculated with the MSMS program [40]. In this work, the values for γ and β were set to 0.00542 kcal mol−1 Å−2 and 0.92 kcal mol−1, respectively, as is standard in the MM-PBSA work that has been published 27.
The contributions of entropy (TΔS) to binding free energy arise from changes of the translational, rotational and vibrational degrees of freedom. TΔS are generally calculated using classical statistical thermodynamics and normal-mode analysis. Due to entropy calculations for large systems being extremely time consuming, we applied only 25 snapshots taken at an interval of 16 ps from the final 400 ps of the MD simulation to calculate the entropy contribution. Each snapshot was minimized with a distance-dependent dielectric function 4R ij (the distance between two atoms) until the root-mean-square of the energy gradient was lower than 10−4 kcal mol−1 Å−2.
MM-PBSA using the single trajectory method usually regards the inhibitor-protein structure as a rigid one and neglects the strain energy produced by the conformational changes, which describe the difference in the bond, angle and dihedral among the complex, the free protease and the free inhibitor [41]. On the other hand, the mutation changes the conformation of the protein in part. Based on the above two reasons, separate MD simulations were also carried out to explore the effect of the conformational changes on the binding. In this separate MD approach, the absolute binding free energy ΔGsep between the inhibitor and the protease was approximately expressed as:
where ΔGcom, ΔGpr and ΔGinh are the free energy of the complex, the free protease and the free inhibitor, respectively. The strain energy involving in the conformational change can be expressed as:
where ΔGstrain, ΔGsep and ΔG are the strain energy, the binding free energy in the separate trajectory and the one in the single trajectory, respectively.
In this work, we also calculated experimental binding free energies from published inhibition constants K i with
where inhibition constants were obtained from Andrey Yu et al. [17].
Because any mutation may result in perturbations that transcend specific localized interactions in the binding cavity, the contributions of individual residue to the binding free energy were calculated by using the MM-GBSA decomposition program in the AMBER 9.0 [29]. The inhibitor-residue interaction is approximated by
where ΔEvdw and ΔEele are non-bonded van der Waals interactions and electrostatic interactions between the inhibitor and each residue in the gas phase. The polar contribution to solvation free energy (ΔGGB) was calculated by using the generalized Born module, and the parameters for the GB calculation were developed by Onufriev et al. [42]. ΔGSUR is the non-polar contribution to solvation free energy and was computed using ΔGSUR = 0.0072 × SA (SA is the accessible surface area). All energy components in Eq. (8) were calculated using 100 snapshots from the last 800 ps of MD simulation.
The hydrogen bonds were analyzed using the ptraj module of AMBER. The formation of the hydrogen bond depended on the distance and the orientation criteria as follow: (1) the distance between proton donor (D) and acceptor (A) atoms was shorter than or equal 3.5 Å, (2) the angle D-H...A was greater or equal to 120°.
Results and discussion
To explore the effect of two mutations on the dynamical stability of the PR/TMC-114 complexes, root-mean-square deviation (RMSD) values for the PR Cα atoms during MD simulation relative to the initial minimized structures were calculated and plotted in Fig. 3. The RMSD values indicate that the conformations of three PR/TMC-114 complexes achieve equilibrium at the same time of about 900 ps after the beginning of MD simulation. According to Fig. 3, RMSD values of the WT, PRD30N/TMC-114 and PRI50V/TMC-114 complexes are 0.87, 0.85 and 0.99 Å, respectively, with a deviation of lower than 0.58 Å from the mean. This result suggests that the stabilities of the dynamic equilibriums for three complexes are reliable.

The root-mean-square deviations (RMSD) of backbone atoms relative to their initial minimized complex structures as function of time: A for the WT, B for the PRI50V and C for the PRD30N
In order to insight into contributions of different components to the binding free energy of TMC-114 to the protease and the effects of two mutants on each component, absolute binding free energies were calculated for three complexes by using MM-PBSA method. Table 1 lists the contributions to the binding free energies calculated from single MD trajectory and separate one, respectively. In the separate trajectory calculations of the binding free energy, the internal energy terms (ΔEint) appears, which is involved in the differences in the bond, angle and dihedral energies among the complex, the free protease and the free inhibitor. As seen from Table 1, the calculations from the single trajectory overestimate the binding affinities, the values of the calculated ΔG are about 10 kcal mol−1 higher than the experimental value, and the rank of ΔG between the PRD30N and the PRI50V is opposite to the one of the experiment (ΔGexp); while the results obtained by using the separate MD simulation approach are closer to the experiment than the ones from the single trajectory, in particular for the PRD30N, moreover, it is so encouraging that the rank of the experimental affinity of the WT, PRD30N and PRI50V/TMC-114 complexes (−13.2, −11.2 and −11.9 kcal mol−1) [17] is consistent with our predictions. Thus, it seemed that the separate MD simulation method is more applicable to the current complexes than the single MD simulation method. The above data suggest that the energetic analysis based on the separate trajectory approach is reliable to the current case study.
According to the energy components of the binding free energy (Table 1), van der Waals and electrostatic terms in the gas phase provide the major favorable contributions to the inhibitor binding. Non-polar solvation energies, which correspond to the burial of SASA upon the binding, also contribute slightly favorably. However, polar solvation energies and entropy terms produce the unfavorable contributions to the binding, the internal energy terms (ΔEint) also oppose the binding. It is observed from Table 1 that the strain energies for the WT, PRD30N and PRI50V /TMC-114 complexes are 7.72, 6.73, and 4.10 kcal mol−1, respectively, which indicates that the mutants D30N and I50V may result in more rigid structure of PR/TMC-114 complex.
From Table 1, the predicted binding free energies of the WT, PRD30N and PRI50V /TMC-114 complexes using the separate MD simulation are −19.06, −14.64, and −16.13 kcal mol−1, respectively. Compared to the WT/TMC-114 complex, the mutants D30N and I50V caused the decrease of −4.42 and −2.93 kcal mol−1 in the affinity, respectively, which suggests that D30N and I50V produce weaker bindings to TMC-114. This also indicates that D30N and I50V provide drug resistance to TMC-114. To explore the mechanism driving the resistance of single mutations D30N and I50V to TMC-114, we compared some independent binding free energy components of PRD30N and PRI50V /TMC-114 complexes with the WT/TMC-114 (Fig. 4). D30N results in a decrease of electrostatic energy by about 8.43 kcal mol−1 relative to the WT, and the decrease in van der Waals energy is about 3.50 kcal mol−1, which drives the resistance of D30N to TMC-114, because the changes of other components do not contribute the reduction in total affinity. In the case of I50V, the main factor driving the resistance to TMC-114 arises from the electrostatic energy, because I50V leads to a decrease of electrostatic energy by about 4.42 kcal mol−1, while van der Waals energy has only a reduction of 0.92 kcal mol−1.

Energy components (kcal mol-1) for the binding of TMC-114 to the WT, PRI50V and PRD30N: (1) electrostatic energy in the gas phase, (2) van der Waals, (3) internal energy (4) non-polar solvation energy, (5) polar solvation energy, (6) entropy
To gain a more-detailed insight into the basis of the resistance of D30N and I50V, the analyses of structure and binding mode were carried out. The binding free energy was decomposed into inhibitor-residue pairs to generate an inhibitor-residue interaction spectrum shown in Fig. 5, which is extremely useful to understand the drug-resistant mechanism of D30N and I50V to TMC-114 at the atomic level. Overall, the interaction spectrums of three complexes are quite similar. The favorable residues mainly come from six groups around Ala28/Ala28′, Ile50/Ile50′, and Ile84/Ile84′. One observation of the energy decomposition spectrum is the binding mode of TMC-114 to chain A of the PR is similar to chain B, although TMC-114 is not a symmetric molecule. Figure 6 shows the decomposition of ΔG values on a per-residue basis into contributions from van der Waals, the sum of electrostatic energy in the gas phase and polar solvation energy, and non-polar solvation energy for residues with |ΔG | ≥ 1.5 kcal mol−1 for three complexes. Table 2 further divides the contributions of per residue into those from backbone atoms and those from side-chain atoms. Figure 7 depicts relative geometries of TMC-114 in the binding complex with relevant residues with which TMC-114 interacts strongly by using the lowest-energy structure from the MD trajectory. The hydrogen bonds were analyzed on the basis of the trajectories with water molecules of the MD simulations (Table 3) to complement the energy analysis.

Decomposition of ΔG on a per-residue basis for the protein-inhibitor complex: (a) the WT/TMC-114, (b) the PRD30N/TMC-114 and (c) the PRI50V/TMC-114

Decomposition of ΔG on a per-residue basis into contributions from the van der Waals energy (vdW), the sum of electrostatic interactions and polar salvation energy (ele+GB) and non-polar solvation energy for the residues of |ΔG| ≥ 1.5 kcal mol-1: (a) the WT/TMC-114, (b) the PRD30N/TMC-114 and (c) the PRI50V/TMC-114

Geometries of eight residues, which produce some major interactions with TMC-114, are plotted in the complex structures determined by the lowest-energy structures from the MD simulation. The hydrogen bonds are shown in the dash line. TMC-114 and Wat301 are indicated in a stick and a ball-and-stick representation. The residues are shown in a line representation
Figure 5a and Table 2 highlight eight residues which contribute most to the binding free energy in the case of the wild-type PR. For all eight residues, the van der Waals energy and non-polar solvation energy drive the binding of TMC-114 to HIV-1 protease (Fig. 6a). The dominating force that drives the binding of TMC-114 to Ala28 is van der Waals energy and the sum of electrostatic energy and polar solvation energy, the total van der Waals energy is −2.34 kcal mol−1 (Table 2), which is produced by the C-H...π interactions between the bis-THF of TMC-114 and the alkyl of Ala28, and the sum of electrostatic energy and polar solvation energy is −1.75 kcal mol−1, which is in agreement with a number of C-H...O interactions formed by the side-chain atoms of Ala28 with the oxygen of the bis-THF and the backbone oxygen of TMC-114 (Fig. 7a). Similar to Ala28, the C-H...π interactions also exist between the aniline of TMC-114 and the alkyl of Ala28′, thus, the total van der Waals energy (−1.80 kcal mol−1) mostly drive the binding of TMC-114 to Ala28′. The N1 atom of TMC-114 forms two hydrogen bonds with Asp30′: one with the backbone nitrogen, another with the side-chain carbonyl oxygen (Table 3 and Fig. 7a). This analysis agrees with the corresponding side-chain electrostatic energy (Sele = −3.08 kcal mol−1) and backbone electrostatic energy (Bele = −3.80 kcal mol−1) in Table 2, and thus the total contributions of electrostatic energy and polar solvation energy is an important driving force for TMC-114 binding to Asp30′ (Fig. 6a). Although the N3 of TMC-114 provide a proton to the carboxylic oxygen of Gly27 to form one hydrogen bond (Table 3 and Fig. 7a), the favorable contribution of this hydrogen bond to the binding is mostly screened by the unfavorable polar solvation energy, thus the hydrophobic interactions, including the van der Waals energy contacts of the C-H...H-C nature between Gly27 and TMC-114, drive the binding of TMC-114 to Gly27. In addition, the central phenyl in TMC-114 contacts the alkyl of the residues Ile50/Ile50′, the aniline of TMC-114 contacts the alkyl of Ile84′ and the bis-THF of TMC-114 also approaches the alkyl of Ile84 to form some donor-π interactions, thus the van der Waals energy favors the binding for these four residues. The above analyses agree generally with the experimental results from Weber et al. [17].
The computed interaction spectrums in Fig. 5b and Fig. 5c show that the PRD30N and PRI50V bind to TMC-114 in similar interaction mode to the WT. The binding analyses of the PRD30N and PRI50V to TMC-114 are also the same as the WT.
In the PRD30N, the aspartate-to-asparagine substitution removes the negative charge of aspartate without significantly changing the geometry of the side chain. Compared to the WT, the PRD30N misses the original hydrogen bond formed by the N1 atom of TMC-114 with the side-chain carbonyl oxygen of Asp30′ in the WT (Table 3 and Fig. 7b), which results in a complete disappearance of the favorable electrostatic energy between TMC-114 and the side chain of Asn30′, and produce an about 0.40 kcal mol−1 repulsive energy (Table 2). Thus, the loss of the hydrogen bond between TMC-114 and the side chain of Asn30′ drive the resistance of D30N to TMC-114. Additionally, Ile84 locates nearby Asn30 (Fig. 7b), and the aspartate-to-asparagine substitution may have a weak influence on the conformation of Ile84, which makes the C-H...π contacts of the alkyls of Ile84 with the aniline of TMC-114 decrease and reduces the van der Waals energy between them (comparing Fig. 6b with Fig. 6a). The reduction in the van der Waals energy between Ile84 and TMC-114, therefore, contributes also slightly to the resistance of D30N to TMC-114.
In the PRI50V, the isoleucine-to-valine substitution reduces the size of the hydrophobic side chain by one methyl group, which is possible to increase the size of the binding cavity and reduce the affinity for inhibitors. The change from the ethyl of isoleucine to the methyl of valine results in a decrease of van der Waals energy between Val50 and TMC-114 by about 0.39 kcal mol−1 relative to the WT (Table 2), because the number of C-H...π contacts between Val50 and the central phenyl of TMC-114 lessen. Although the Ile50′ mutation to Val50′ produce a dramatic increase of electrostatic energy in the gas phase between TMC-114 and Val50′ in comparison to the WT, this favorable effect is completely counteracted by the positive solvation energy, which suggests the polar solvation energy is an essential contributor to the resistance of I50V to TMC-114 (Table 2 and Fig. 6c). Additionally, two hydrogen bonds exist between TMC-114 and Asp30′: one is aniline of the inhibitor with the backbone nitrogen of Asp30′, another for aniline with the side-chain carbonyl oxygen (Table 3 and Fig. 7c), which is in agreement with the favorable electrostatic interaction of TMC-114 with the backbone and side chain of Asp30′ (Table 2), but the favorable electrostatic interaction in the gas phase is offset by the polar solvation energy (Fig. 6c). Based on the energy and structure analysis above, the conclusion can be made that the increase in the polar solvation energy between TMC-114 and two residues Val50′ and Asp30′ definitively drives the resistance of I50V to TMC-114.
In addition, the water molecule Wat301 appears in the three studied complex, and it links the SO2 and carbonyl of the urethane moiety with the main-chain amides of residues Ile50 (Val50) and Ile50′ (Val50′) by means of four hydrogen bonds (Table 3 and Fig. 7). According to Table 3, the occupancy of these four hydrogen bonds is higher than 90%, in despite of the isoleucine-to-valine substitution directly relating with these hydrogen bonds in the PRI50V, which suggests that D30N and I50V do not produce the influence on the stability and function of Wat301. This result indicates that the water molecule Wat301 hardly contribute the resistance of D30N and I50V to TMC-114. The above conclusions concerning mutations are in accord with the experimental analysis generally [17].
Conclusions
In this work, 3-ns MD simulations combined with the calculation of binding free energy by using the MM-PB/SA method were carried out to analyze the drug resistance of D30N and I50V to TMC-114, and also investigate the binding mechanisms of TMC-114 to the WT, PRD30N, and PRI50V. The results suggest that the decrease in the van der Waals energy and electrostatic energy in the gas phase definitively produce the drug resistance of D30N to TMC-114, while for I50V, the decrease in the electrostatic energy mainly drive its drug resistance to TMC-114. The separate MD simulations of the complex, the protein and the inhibitor were performed to investigate the effects of the conformational changes on the binding. The computed strain energies show that D30N and I50V result in a more rigid structure of the PR/TMC-114 complex than the WT.
The analyses of the structure-affinity relationship were applied to investigate the binding mode of TMC-114 to the PR and the drug-resistance mechanisms of D30N and I50V to TMC-114. Among three complexes, the favorable interactions come from Gly27, Ala28/Ala28′, Asp30′(Asn30′), Ile50/Ile50′ or Val50/Val50′ and Ile84/Ile84′. The favorable interactions mainly were produced by four types of interactions: the hydrogen bond interaction, the C-H...π interactions, the C-H...O interactions and the C-H...H-C interactions. The comparisons of the structure-affinity relationship between the PR and mutant PR expose the resistant mechanisms of two mutations to TMC-114. The loss of the hydrogen bond between TMC-114 and the side chain of Asn30′ is the main driving force of the resistance of D30N to TMC-114, additionally, the reduction in the van der Waals energy between Ile84 and TMC-114, also contributes slightly to the resistance of D30N to TMC-114. For I50V, the increase in the polar solvation energies between TMC-114 and two residues Val50′ and Asp30′ definitively drives the resistance of I50V to TMC-114. The analyses of the hydrogen bonds concerning the water molecule Wat301 were done to investigate the effect of Wat301 on the resistance, and the data show that Wat301 hardly contribute the resistance of D30N and I50V to TMC-114.
This article provides a quantitative and mechanistic explanation of mutational effect from detailed analyses of the structure-affinity relationship. We expect that this work can provide some helpful insights into the nature of mutational effect and aid the future design of more potent inhibitors.
References
Navia MA, Fitzgerald PMD, Mckeever BM, Leu CT, Heimbach JC, Herber WK, Sigal IS, Darke PL Springer JP (1989) Nature 337:615–620
Perryman AL, Lin JH, Andrew McCammon J (2006) Biopolymers 82:272–284
Roberts NA, Martin JA, Kinchington D, Broadhurst AV, Craig JC, Duncan IB, Galpin SA, Handa BK, Kay J, Krohn A et al (1990) Science 248:358–361
Kempf DJ, Marsh KC, Denissen JF, McDonald E, Vasavanonda S, Flentge CA, Green BE, Park FL, CH KXP et al (1995) Proc Natl Acad Sci USA 92:2484–2488
Vacca JP, Dorsey BD, Schleif WA, Levin RB, McDAniel SL, Darke PL, Zugay J, Quintero JC, Blahy OM, Roth E et al (1994) Proc Natl Acad Sci USA 91:4096–4100
Kalish V, Kaldor S, Shetty B, Tatlock B, Davies J, Hammond M, Dressman B, Fritz J, Appelt K, Keich S, Su K (1995) Eur J Med Chem 30:201s–214s
Reddy P, Ross J (1999) Formular 18:567–577
Sham HL, Kempf DJ, Molla A, Marsh K, Kumar GN, Chen CM, Kati W, Stewart K, Lal R, Hsu A (1998) Antimicrob Agents Chemother 42:3218–3224
Bold G, Fassler A, Capraro HG, Cozens R, Klimkait T, Lazdins J, Mestan J, Poncioni B, Rosel J, Stover D, Tintelnot-Blomley M, Acemoglu F, Beck W, Boss E, Eschbach M, Hurlimann T, Masso E, Roussel S, Ucci-Stoll K, Wyss D, Lang M (1998) J Med Chem 41:3387–3401
Sommadossi JP (1998) Antivir Ther 3:9–12
Flexner C (1998) N Engl J Med 338:1281–1292
Moyle G (2002) AIDS Patient Care STDS 16:585–597
Hertogs K, Bloor S, Kemp SD, Van den Eynde C, Alcorn TM, Pauwels R, Van Houtte M, Staszewski S, Miller V, Larder BA (2000) AIDS 14:1203–1210
Wu TD, Schiffer CA, Gonzales MJ, Taylor J, Kantor R, Chou S, Israelski D, Zolopa AR, Fessel WJ, Shafer RW (2003) J Virol 77:4836–4847
Ohtaka H, Freire E (2005) Prog Biophys Mol Biol 88:193–208
Tie Y, Boross PI, Wang Y-F, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT (2004) J Mol Biol 338:341–352
Kovalevsky AY, Tie Y, Liu F, Boross PI, Wang YF, Leshchenko S, Ghosh AK, Harrison RW, Weber IT (2006) J Med Chem 49:1379–1387
Nivesanond K, Peeters A, Lamoen D, Alsenoy CV (2008) J Chem Inf Model 48:99–108
Jarvis B, Faulds D (1998) Drugs 56:147–167
Wu EL, Han KL, Zhang JZH (2008) Chem Eur J 14:8704–8714
Hou T, Zhang W, Case DA, Wang W (2008) J Mol Biol 76:1201–1214
Wu EL, Mei Y, Han KL, Zhang JZH (2007) Biophys J 92:4244–4253
Xu Y, Wang R (2006) PROTEINS 64:1058–1068
Strockbine B, Rizzo RC (2007) PROTEINS 67:630–642
Zou H, Luo C, Zheng S, Luo X, Zhu W, Chen K, Shen J, Jiang H (2007) J Phys Chem B 111:9104–9113
Wang W, Kollman PA (2001) PNAS 26:14939–14942
Wang J, Morin P, Wang W, Kollman PA (2001) J Am Chem Soc 123:5221–5230
Srinivasan J, Millar J, Kollman PA, Case DA (1998) J Biomol Struct Dyn 16:265–280
Gohlke H, Kiel C, Case DA (2003) J Mol Biol 330:891–913
Hyland LJ, Tomaszek TA, Meek TD (1991) Biochem-US 30:8454–8463
Hou T, Mclaughlin WA, Wang W (2008) PROTEINS 71:1163–1174
Hou T, Yu R (2007) J Med Chem 50:1177–1188
Case DA, Darden TA, Cheatham TE III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WS, Kollman PA (2006) AMBER 9. University California, San Francisco
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski JW, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Gonzalez C, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA (1998) Gaussian 98. Gaussian Inc, Pittsburgh, PA
Bayly CI, Cieplak P, Cornell WD, Kollman PA (1995) J Phys Chem 97:1367–1377
Wang JM, Gieplak P, Kollman PA (2000) J Comput Chem 21:1049–1074
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1998) J Chem Phys 79:926–935
Darden T, York D, Pedersen L (1993) J Chem Phys 98:10089–10092
Ryckaert JP, Ciccotti G, Berendsen HJC (1977) J Comput Phys 23:327–341
Sanner MF, Olson AJ, Spehner J (1996) Biopolymers 38:305–320
Lepsík M, Kríz Z, Havlas Z (2004) PROTEINS 57:279–293
Onufriev A, Bashford D, Case DA (2000) J Phys Chem 104:3712–3720
Acknowledgments
This work is supported by the National Nature Science Foundation of China (Grant Nos. 10874104, 10474060 and 10504017). The key Project of Chinese Ministry of Education (NO. 206093), the key Project of Nature Science Foundation of Shandong Province (Z2007A05) and Shandong Province Foundation for Middle-aged and Young Scientist (2008BS01013).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chen, J., Zhang, S., Liu, X. et al. Insights into drug resistance of mutations D30N and I50V to HIV-1 protease inhibitor TMC-114: Free energy calculation and molecular dynamic simulation. J Mol Model 16, 459–468 (2010). https://doi.org/10.1007/s00894-009-0553-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-009-0553-7