
Abstract
“Halomonas socia” NY-011, a new species of moderately halophilic bacteria isolated and identified in our laboratory, can grow in high concentrations of salt ranging from 0.5 to 25%. In this study, the whole genome of NY-011 was sequenced and a detailed analysis of the genomic features was provided. Especially, a series of genes related to salt tolerance and involved in xenobiotics biodegradation were annotated by COG, GO and KEGG analyses. Subsequently, RNA-Seq-based transcriptome analysis was applied to explore the osmotic regulation of NY-011 subjected to high salt stress for different times (0 h, 1 h, 3 h, 6 h, 11 h, 15 h). And we found that the genes related to osmoregulation including excluding Na+ and accumulating K+ as well as the synthesis of compatible solutes (alanine, glutamate, ectoine, hydroxyectoine and glycine betaine) were up-regulated, while the genes involved in the degradation of organic compounds were basically down-regulated during the whole process. Specifically, the expression trend of genes related to osmoregulation increased firstly then dropped, which was almost opposite to that of degrading organic pollutants genes. With the prolongation of osmotic up-shock, NY-011 survived and gradually adapted to osmotic stress, the above-mentioned two classes of genes slowly returned to normal expression level. Then, the scanning electron microscope (SEM) and transmission electron microscope (TEM) were also utilized to observe morphological properties of NY-011 under hypersaline stress, and our findings showed that the cell length of NY-011 became longer under osmotic stress, at the same time, polyhydroxyalkanoates (PHAs) were synthesized in the cells. Besides, physiological experiments confirmed that NY-011 could degrade organic compounds in a high salt environment. These data not only provide valuable insights into the mechanism of osmotic regulation of NY-011; but also make it possible for NY-011 to be exploited for biotechnological applications such as degrading organic pollutants in a hypersaline environment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In this era of uncurbed industrialization around the world, industrial effluents have increased more sharply than ever before. Often, they are hypersaline and frequently contaminated with organic compounds (Oren et al. 1992). Besides, hypersaline wastewater with massive of organic compounds and various pollutants may be produced in many fields such as papermaking, agriculture, petroleum, printing and dyeing, pharmaceutical and other chemical industries (Díaz et al. 2002; Lefebvre et al. 2005). These industrial saline effluents are more harmful and more difficult to deal with, and the direct discharge of them without pre-treatment will have a greater impact on the surrounding soil, water environment, agricultural production and human health (Lefebvre and Moletta 2006). Especially polycyclic aromatic hydrocarbons (PAHs), generated by the petroleum industry, poses serious threats to environmental and human health due to their carcinogenicity, teratogenicity, genotoxicity (Kim et al. 2013).
At present, many methods for the treatment of high-salt wastewater with organic pollutants are provided, both physical and chemical, but most of them are associated with higher treatment costs. Additionally, some of them may generate noxious by-products and cause secondary pollution (Ahmadi et al. 2017; Zhao et al. 2020). However, the biological method has the unique advantages of low-cost and no harmful by-products produced in the treatment process using various microorganisms to degrade organic pollutants, which has aroused great interest of researchers (Mirbolooki et al. 2017). Nevertheless, traditional microbes cannot survive and degrade pollutants efficiently in the condition of high salinity. They cannot cope with hypersaline stress which would disrupt their cellular osmotic balance, disturb the enzymatic reaction, break the integrity of the cell membrane and affect the normal physiological metabolism process (Pradhan et al. 2016). Therefore, desalination or dilution to appropriate salt concentration is often used as pretreatment of bioremediation at present, which complicates the process and increases the cost, on the other hand, it also causes a lot of waste of freshwater resources. But using moderately halophilic bacteria that are able to survive under high salinity can overcome these limitations, thus being the potential candidates for degrading the pollutants at high salt environments (Ceylan et al. 2011).
Many pieces of literature have reported that moderately halophilic bacteria (especially Halomonas) can degrade a variety of organic pollutants, including polycyclic aromatic hydrocarbons, azo dyes, phenols, hydrocarbons in crude oil and so on. For example, Govarthanan et al. (2020) isolated a halophilic bacteria Halomonas sp. RM from brackish water which was demonstrated to biodegrade phenanthrene (Phe), pyrene (Pyr), naphthalene (NaP) and benzo [a] pyrene (BaP) in a wide range of pH, carbon sources and salinity. Amini et al. (2019) reported that the maximum biodegradation of azo dye Toluidine Red (C.I. no.12120) by Halomonas strain Gb was 81%. Haddadi and Shavandi (2013) isolated a new highly efficient phenol-degrading bacteria Halomonas sp. PH2-2 can tolerate a high concentration of salt and phenol from high saline soil. Mnif et al. (2009) isolated a salt-tolerant efficiently hydrocarbons-degrading bacteria Halomonas sp. C2SS100 from production water of an oil field after enrichment on crude oil.
However, at present, most of the studies about biodegradation of organic pollutants in saline conditions are still focused on the screening and identification of halophilic microorganisms which can degrade organic compounds in the high-salt environment. The metabolic process of biodegrading various organic pollutants by halophilic microorganisms under a high saline environment is rarely studied. Thereby, to understand the mechanism of biodegradation of organic compounds by halophiles under hypersaline environment is of great significance and potential value for developing effective bioremediation strategies under saline conditions.
As a new species of moderately halophilic bacteria isolated and identified in our laboratory (Cao et al. 2013), “Halomonas socia” NY-011 possesses unusual capacities of robustness to stress, degradation of organic compounds and wide substrate adaptability, having a great potential application prospect of biotechnology. So High-throughput sequencing for genomics and transcriptomics of strain NY-011, as well as scanning electron microscope (SEM), transmission electron microscope (TEM) and physiological experiments were performed in this study, hoping to provide meaningful value about (i) the osmoregulation mechanism of halophilic microorganism and (ii) the metabolic process of degrading organic compounds under high saline environment, which could be an important part for developing bioremediation technology under saline conditions.
Materials and methods
Genome sequencing and annotation
“Halomonas socia” NY-011 [preserved in China Center for Type Culture Collection (CCTCC AB 2,011,033) and Korean Collection for Type Cultures (KCTC 23,671)] was inoculated into test tube containing 5 ml 8% NaCl LB liquid medium [8% NaCl (w/v), 1% tryptone (w/v) and 0.5% yeast extract (w/v)] with an orbital agitation of 200 r/min, at 30 ℃ until the mid-logarithmic phase. Its genomic DNA was extracted using TIANamp Bacteria DNA Kit following manufactures protocol (TIANGEN Biotech Company, Beijing, China), and then sequenced at Genewiz Biotechnology Co. Ltd. (Suzhou, China). The specific procedure was as follows: Genomic DNA was sheared, then 9 Kb double-stranded DNA fragments were selected. DNA fragments were end-repaired and ligated with universal hairpin adapters. Subsequent steps were followed as per the manufacture’s instruction to prepare the SMRTbell library. The library was sequenced in the PacBio RSII instrument (Mccarthy 2010), then the PacBio reads were assembled using Falcon of WGS-Assembler 8.2 (Berlin et al. 2015). And software Quiver (working with Pacbio reads) was used to recorrect the genome. Consequently, a genomic fine drawing was obtained by mapping PacBio data to the assembled genome using BWA software (version 0.7.12) (Li and Durbin 2009, 2010). Protein coding sequence was predicted by Glimmer software (version 3.0) (Delcher et al. 2007) and annotated using BLAST to search for non-redundant protein sequences from the NR, Swiss-Prot (Bairoch and Apweiler 2000), Pfam (Finn et al. 2014), EggNOG (Jensen et al. 2007), GO and KEGG (Kanehisa and Goto 2000) databases. Genes for transfer RNA (tRNAs) were detected using tRNAscan-SE software version 2.0 (Besemer and Borodovsky 2005) and genes encoding ribosomal RNA (rRNAs) were predicted using the RNAmmer software 1.2 (Lagesen et al. 2007). Tandem repeat sequences were predicted using Tandem Repeats Finder software (Benson 1999). The genomic data have been deposited in the GenBank with the accession number CP049827.
Extraction and determination of intracellular solutes by nuclear magnetic resonance (NMR) spectroscopy
13C nuclear magnetic resonance (NMR) analysis was used to identify compatible solutes in NY-011. Extraction and determination of intracellular solutes using NMR spectroscopy described by Cánovas et al. 1999.
Reverse transcription quantitative PCR (RT-qPCR)
Total RNA was extracted from NY-011 using TRIzol® Reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. Reverse transcription was performed using the PrimeScript™ RT reagent kit with gDNA Eraser (Takara Biotechnology Co., Ltd., Japan). qPCR was conducted using an SYBR® Premix Ex Taq™ II (Takara Biotechnology Co., Ltd., Japan) following the manufacturer’s protocol in the Bio-Rad CFX 96 Real-time Detection System (Bio-Rad). Primers for genes ectA (ectA-F: 5′-ATTCCGCCTACGCCTATCT-3′ and ectA-R: 5′-AGGGGCGTTGCTCTTCAC-3′) and 16S rDNA (16S rDNA-F: 5′-TCAGGCTAGAGTGCAGGAGAG-3′ and 16S rDNA-R: 5′-TCGTTTACGGCGTGGACTA-3′) were synthesized by Tsingke Biological Technology Co. (Chengdu, China). The relative quantification of the gene was determined by calculating the values of 2−ΔΔCt. In the present study, 16S rRNA was used as the reference gene for the normalization of gene ectA. The experiment was repeated in triplicate.
Transcriptome sequencing and analyses
NY-011 was incubated in LB medium containing 8% NaCl (w/v) until the mid-logarithmic phase firstly. Subsequently, 5 ml suspensions of fresh cells were collected by centrifugation at 6000 r/min for 10 min and then inoculated into 5 ml LB medium containing 15% NaCl (w/v). Six bacterial samples named A0, A1, A3, A6, A11 and A15 (each sample is in triplicates) incubated for 0 h, 1 h, 3 h, 6 h, 11 h and 15 h, respectively, finally a sample in triplicates was mixed into one sample. Total RNA of each mixed sample was extracted using TRIzol® Reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. The rRNA was depleted from total RNA using the Ribo-Zero rRNA Removal Kit (Bacteria) (Illumina). Next-generation sequencing library preparations were constructed according to the manufacturer’s protocol (NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina®). Then libraries were loaded on an Illumina HiSeq instrument according to manufacturer’s instructions (Illumina, San Diego, CA, USA). Sequencing was carried out using a 2 × 150 paired-end (PE) configuration; image analysis and base calling were conducted by the HiSeq Control Software (HCS) + OLB + GAPipeline (Illumina) on the HiSeq instrument. The sequences were processed and analyzed by Genewiz Biotechnology Co. Ltd. (Suzhou, China). FPKM (expected number of Fragments Per Kilobase of transcript sequence per Millions of base pairs sequenced) was calculated by Htseq (V 0.6.1) to analyze gene expression of unigenes. Differentially Expressed Genes (DEGs) were identified with FDR (False Discovery Rate) ≤ 0.05 and FPKM > 1, and Significantly Differentially Expressed Genes (SDEGs) were identified with FDR ≤ 0.05, FC (Fold Change of differential gene expression) ≥ 2 and FPKM > 1 (Zhang et al. 2018). The RNA-Seq data have been deposited in the NCBI Sequence Read Archive (SRA) with the accession number SRR11234929 for A0, SRR11234928 for A1, SRR11234927 for A3, SRR11234926 for A6, SRR11234925 for A11 and SRR11234924 for A15.
Bacteria morphology observation
Bacterial cultivation and high-salt treatment were performed as a previous description in transcriptome sequencing and analyses. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) of bacteria samples were prepared as described by Tan et al. (2017). Briefly, the samples of NY-011 for SEM were fixed with 2.5% glutaraldehyde, dehydrated with ethanol and tertiary butanol, then dried with CO2, coated with gold–palladium and examined using a JEOL JSM-7500F (JEOL, Tokyo, Japan). For transmission electron microscopy (TEM), the samples were prefixed with 2.5% glutaraldehyde, then post-fixed with 2% osmium tetroxide and dehydrated in a graded ethanol series. Then, the specimens were incubated with epoxy resin and subsequently sectioned using an ultramicrotome, finally stained with uranyl acetate and lead citrate, and observed using a Hitachi HT7700 (Hitachi, Tokyo, Japan).
Pyrene tolerance and biodegradation experiments
To study the tolerance of pyrene by the “Halomonas socia” NY-11, the cells were inoculated into 100 ml flasks containing 30 ml 8% NaCl 1/2 LB liquid medium [8% NaCl (w/v), 0.5% tryptone (w/v) and 0.25% yeast extract (w/v) added different concentrations of pyrene at a final concentration of 0 mg/L, 10 mg/L, 50 mg/L, 100 mg/L, 150 mg/L, 200 mg/L, 300 mg/L, 400 mg/L and 500 mg/L with an orbital agitation of 200 r/min, at 30 ℃. Each experiment was conducted with three biological replicates. Samples were taken and measured using Optical Density at 600 nm (OD600) by ultraviolet spectrophotometer at 6 h intervals until the cultures reached their plateau phases.
Then, to study the biodegradation of pyrene by the “Halomonas socia” NY-11, pyrene was added into 100 ml flasks containing 30 ml 8% NaCl LB liquid medium at a final concentration of 100 mg/L with NY-011 inoculated as an experimental group and without an inoculum as a control. Each experiment was conducted at an orbital agitation of 200 r/min and 30 ℃ with three biological replicates. Samples were taken every 7 days for one month and residual pyrene of each sample was extracted by liquid–liquid extraction using ethyl acetate as a solvent, and analyzed by GC–MS. More details were described by Nzila et al. (2018).
Results
Complete genome analyses
Genome features
After mapping PacBio data to the assembled genome, 99.99% coverage was achieved, Mean Depth was 164.73, and a complete genome sequence with a total of 4,780,349 base pairs was obtained. The complete genome of “Halomonas socia” NY-011 is composed of only one 4.78 Mb circular chromosome without a plasmid. A total of 4581 genes are predicted in the whole genome. The average length of genes is 930.37 bp, and the total coding gene sequences account for 89.16% in the whole genome. The average GC content in the gene region is 65.05%, which is higher than the GC content of the whole genome (64.20%), and the average GC content in the intergenic region is only 57.19%. Also, there are 100 non-coding RNAs predicted, including 12 rRNAs, 66 tRNAs and 22 other non-coding RNAs. Besides, 70 tandem repeats with total lengths of 12,401 bp obtained in “Halomonas socia” NY-011 accounting for 0.29% of the total length of the genome. All information is summarized in Table 1.
COG analyses
By searching against the EggNOG database, we found that a total of 4055 genes of “Halomonas socia” NY-011 were annotated accounting for 88.52% of all genes. These annotated genes were classified into 22 categories (Fig. 1). Among them, 1066 genes were assigned to Functional Unknown (S) the number of which was the largest. Secondly, there were 398, 333, 309 genes annotated to Amino Acid Transport and Metabolism (E), Inorganic Ion Transport and Metabolism (P), Transcription (K), respectively. And there were 262, 260, 222 genes annotated to Energy Production and Conversion (C), Carbohydrate Transport and Metabolism (G), Replication, Recombination and Repair (L), respectively. The number of genes assigned to the remaining functional categories is less than 200.

The COG function classification of strain NY-011
Gene ontology (GO) analyses
A blast search against the GO database showed that a total of 3225 genes were annotated making up 70.4% of all genes, and 2499 genes were annotated to “Molecular Function”, 2405 genes annotated to “Biological Process”, 1568 genes annotated to “Cellular Component” (Fig. 2). It is worth noting that although there were the least genes annotated to “Cellular Component”, its secondary category “components of the membrane” was enriched the most of genes (765 genes) which is far more than that of other secondary categories. Besides, many genes were also annotated to other membrane-related secondary classifications, such as “plasma membrane” (240 genes), “membrane” (137 genes), “outer membrane space” (56 genes), “outer membrane” (24 genes) and “components of plasma membrane” (19 genes).

The GO annotation of strain NY-011
KEGG enrichment analyse
To identify the biological pathways of NY-011, we mapped all genes to reference canonical pathways in the KEGG database. A total of 2548 genes were divided into six categories: Cellular processes (217 genes), metabolism (1597 genes), human diseases (121 genes), genetic information processing (199 genes), organismal systems (36 genes), and environmental information processing (378 genes) (Fig. 3).

The KEGG annotation of strain NY-011
Genes related to osmotic regulation
By analyzing the whole genome sequence of NY-011, genes related to the synthesis of ectoine, hydroxyectoine, glycine betaine, glutamate and alanine were identified, and genes involved in potassium ion and sodium ion transport were also identified. All information is shown in Table 2.
13C NMR analysis of the compatible solutes in NY-011
The chemical shift δ (ppm) values were compared with the published standard spectrum to identify the types of compatible solutes in NY-011. The ppm values of alanine are 1.69, 50.9, 176.2; ppm values of glutamate are 27.5, 33.5, 55.0, 174.5, 181.4; ppm values of ectoine are 17.9, 23.6, 38.5, 54.8, 161.9, 177.8; ppm values of hydroxyectoine are 18.7, 45.2, 69.8, 162.9, 175.5; ppm values of glycine betaine are 54.2, 66.9, 170 (Cánovas et al.1999 , 1997 ). We found that NY-011 can synthesize five compatible solutes (E: ectoine; B: glycine betaine; G: glutamate; H: hydroxyectoine; A: alanine), mainly ectoine, glycine betaine and hydroxyectoine (which exist at all tested salt concentrations) (Fig. 4a–e).

13C NMR analysis of the compatible solutes in NY-011 at different NaCl concentrations. Pictures (a–e) represent compatible solutes in NY-011 at 5%, 10%, 15%, 20% and 25% NaCl, respectively. E ectoine, B glycine betaine, G glutamate, H hydroxyectoine, A alanine
RT-qPCR detecting the expression of ectA
According to the results of 13C NMR analysis, ectoine is one of the main compatible solutes synthesized by NY-011. Therefore, to explore the differential transcription of ectA gene (ectoine synthesis gene ectABC are reported to form an operon) at a different time under hypersaline environment, we detected the gene expression of ectA by Quantitative Real-time PCR after NY-011 was subjected to high salt stress of 15% NaCl for 0 h, 1 h, 3 h, 5 h, 7 h, 9 h, 11 h, 13 h, 15 h, 17 h respectively. We found that transcripts of the ectA gene increased dramatically first and reached a maximum at 1 h after the osmotic upshock, then began to decrease and reached a minimum at 11 h, and then increased and reached the second peak at 15 h.
Transcriptome analyses
Screening of significantly differentially expressed genes
According to the results of differential expression of ectA at different time, “Halomonas socia” NY-011 was subjected to hyperosmotic stress at 15% NaCl for 0 h, 1 h, 3 h, 6 h, 11 h and 15 h, respectively, and then sent to the company for transcriptional sequence. Among them, the sample of 0 h (named A0) was taken as the control group, while the samples of 1 h, 3 h, 6 h, 11 h and 15 h (named A1, A3, A6, A11, A15, respectively) were taken as the experimental groups, and a total of 5 groups (A0-VS-A1, A0-VS-A3, A0-VS-A6, A0-VS-A11 and A0-VS-A15) were obtained. Significantly differentially expressed genes (SDEGs) were screened according to log2|FC| ≥ 1 and FDR ≤ 0.05 (fold change of differential gene expression ≥ 2 and False Discovery Rate ≤ 0.05). Group A0-VS-A1, A0-VS-A3, A0-VS-A6, A0-VS-A11 and A0-VS-A15 totally acquired SDEGs of 839 (down-regulation: 535; up-regulation: 304), 493 (down-regulation: 341; up-regulation: 152), 235 (down-regulation: 157; up-regulation: 78), 380 (down-regulation: 169; up-regulation: 211) and 490 (down-regulation: 211; up-regulation: 279), respectively (Fig. S1).
SDEGs related to xenobiotics biodegradation
We analyzed the A0-VS-A1 group firstly because of the number of SDEGs in this group is being the largest. The top 15 significantly enriched metabolic pathways in the A0-VS-A1 group were summarized in Table S1 by mapping all SDEGs to reference canonical pathways in the KEGG database. We found that most of these metabolic pathways are involved in xenobiotics biodegradation, and SDEGs enriched in the pathways are all significantly down-regulated. Combined with results of genome and transcriptome analyses, as well as harmfulness, universality, degradation cycle of related xenobiotics, four pollutants and their metabolic pathways (Degradation of aromatic compounds, Atrazine degradation, Toluene degradation and Styrene degradation) were selected for in-depth analysis.
A total of 22 SDEGs were significantly enriched in the four metabolic pathways: Degradation of aromatic compounds (11 SDEGs), Atrazine degradation (3 SDEGs), Toluene degradation (4 SDEGs), Styrene degradation (4 SDEGs). These genes encode S-(hydroxymethyl) glutathione dehydrogenase, anthranilate dioxygenase reductase, benzene 1,2-dioxygenase, anthranilate 1,2-dioxygenase, catechol 1,2-dioxygenase, homogentisate 1,2-dioxygenase and so on (Table 3). We also realized that at high salt conditions the SDEGs for the mentioned pathways were significantly down-regulated at the beginning (0–3 h), but expression increased over time of salt exposure (3–15 h) (Fig. 5a–d).


Genes related to xenobiotics degradation and osmotic regulation as well as their expression trend. The expression trend of genes involved in the aromatic compound degradation pathway (a), atrazine degradation pathway (b), toluene degradation pathway (c), styrene degradation pathway (d), ectoine synthesis (e), glycine betaine metabolism (f), glutamate metabolism (g), alanine metabolism (h), primary sodium pump (i), Na+/H+ antiporter (j) and K+ transport system (k)
Differentially expressed genes (DEGs) related to osmotic regulation
DEGs related to ectoin
DEGs related to ectoine and hydroxyectoine synthesis were up-regulated in NY-011 during the whole osmotic upshock (Fig. 5e). Ectoine synthesis genes (ectABC) were all up-regulated by more than twofold while the hydroxyectoine synthesis gene (ectD) was merely up-regulated by 1.41-fold when NY-011 was treated with hypersaline stress in the first hour. With the extension of treating time, ectABC was still up-regulated, but their expression dropped, while the expression of ectD still increased until the eleventh hour then dropped.
DEGs related to glycine betaine
Subjected to high salt stress for 1 h, the genes (betA and betB) encoding choline dehydrogenase and betaine-aldehyde dehydrogenase related to glycine betaine synthesis in NY-011 were significantly up-regulated by 16.11-fold and 14.95-fold, respectively. When treated for 3 h, betA and betB were further significantly up-regulated by 33.66-fold and 30.24-fold, respectively. With the extension of treating time, betA and betB were still up-regulated, but their expression decreased (Fig. 5f).
DEGs related to glutamate
Under the hypersaline condition, the expression trend of the gene (putA) encoding proline dehydrogenase which can convert l-1-pyrroline-5-carboxylic acid to l-glutamate, was consistent with that of betA and betB. while the glutamate degradation gene glnA, the product of which can convert l-glutamate to l-glutamine, was significantly down-regulated all the time. Besides, gdhA and gudB encode two kinds of glutamate dehydrogenase, respectively, which can catalyze the mutual conversion between 2-ketoglutarate and l-glutamic acid. However, gdhA was always up-regulated which was almost opposite to gudB (Fig. 5g).
DEGs related to alanine
Alanine aminotransferase encoded by gene alt can convert pyruvate to l-alanine, and alanine-glyoxylate transaminase encoded by gene agxT2 can catalyze the degradation of l-alanine. Under hypersaline stress, agxT2 was down-regulated at 1 h and then up-regulated, while the expression trend of alt was almost opposite to that of agxT2. Alanine dehydrogenase encoded by gene ald can catalyze the conversion of pyruvate to l-alanine. However, none of them reached a significant differential expression (Fig. 5h).
DEGs related to Na+ transport system
Under salt stress, the expression trend of primary sodium pump genes nqrABCD increased at first and then dropped. And these four genes were significantly up-regulated with their transcripts reaching a maximum at 1 h after the osmotic upshock (Fig. 5i). The genes related to Na+/H+ antiporter including mrpA_2, mrpB_1, mrpC_2, mrpD_2, mrpE, mrpF_2, mrpG_2, etc. were up-regulated within 0 ~ 6 h, and their expression did not reach a significant differential expression level (Fig. 5j).
DEGs related to K+ transport system
The genes involved in K+ transport system were all down-regulated at 1 h, then up-regulated, and further up-regulated with the prolongation of stress time (Fig. 5k).
Morphological observation of NY-011 under high salt stress
In this study, an optical microscope, scanning electron microscope (SEM) and transmission electron microscope (TEM) were used to investigate the morphological changes of NY-011 under osmotic upshock at different time.
Results of optical microscope observation
After NY-011 being subjected to high salt stress of 15% NaCl for 0 h, 0.5 h, 1 h, 1.5 h, 2 h, 2.5 h, 3 h, 4 h, 5 h, 6 h, 9 h, 12 h and 15 h, respectively, we observed that the average length of NY-011 cells increased from 0 to 3 h, and gradually decreased to the minimum at 15 h. Thus the cells stressed for 0 h, 1 h, 3 h, 6 h and 15 h were collected for further scanning electron microscope observation.
Results of scanning electron microscope observation
No cell membrane shrinkage or cell damage and cell fragments caused by osmotic shock were observed from scanning electron microscope. In the observation, we also found the same phenomenon that the average length of NY-011 cells increased from 0–3 h, and gradually decreased to the minimum at 15 h as the results of optical microscope observation (Fig. 6). Besides, all cells covering with a bright membrane (shown by the orange arrow in Fig. 6e) are clustered together which is difficult to find a single scattered cell. And the flocs can be seen next to the cells (shown by the red arrow in Fig. 6a). Obvious cracks which can be formed by the cracking of organic substances after dehydration and drying can also be observed between cells (shown in the red circle in Fig. 6c, e). All these phenomena may be caused by the presence of a bacterial capsule which can help bacteria resist the adverse environment. Combined with results of optical microscope and scanning electron microscope observation, NY-011 cells stressed for 0 h (control group) and 3 h (experimental group) were collected for further transmission electron microscope observation due to their biggest difference on the cell length.

Cell morphology of strain NY-011 under a scanning electron microscope. Pictures (a–e) represent strain NY-011 subjected to 15% NaCl for 0 h, 1 h, 3 h, 6 h, 15 h, respectively, and picture (f) means changes of the average length of strain NY-011 under different stress time
Results of transmission electron microscope observation
NY-011 cells in the control group and the experimental group are all relatively complete without obvious rupture or cell fragments. Besides, in the control group, the outer membrane was irregular with large surface area, and part of which protruded and formed vesicles, while the inner membrane was relatively smooth. And there was a large periplasmic space between the inner and outer membranes. Besides, PHA-like granules can be seen in the cells. Compared with the control group, the outer membrane of cells in the experimental group is more regular with more outer membrane protruding to form vesicles. And there are also many vesicles between the cells. The periplasmic space between the inner and outer membrane is smaller. And PHA-like granules are also present in the cells (Fig. 7).

Morphology and internal structure of strain NY-011 under transmission electron microscope Pictures (a, b) represent control group with a resolution of 2.0 μm and 500 nm, respectively; pictures (c, d) represent an experimental group (strain NY-011 subjected to 15% NaCl for 3 h) with a resolution of 2.0 μm and 500 nm, respectively. Red arrows 1–5 represent vesicle, PHA-like granule, inner membrane, outer membrane and periplasmic space, respectively
Verification of xenobiotics degradation by NY-011
Tolerance of NY-011 to pyrene
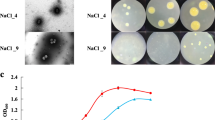
Our studies showed that NY-011 could not utilize toluene, styrene, atrazine, polycyclic aromatic hydrocarbons (naphthalene, phenanthrene, anthracene, fluoranthene, pyrene) as the only carbon source and energy source for growth and reproduction excepted LB medium present even a small amount of it. And compared with other organic compounds, the strain grew best in LB medium added pyrene. Therefore, in this experiment, NY-011 was transferred into 1/2 LB liquid medium containing different concentrations of pyrene at 8% NaCl to explore the tolerance of NY-011 to pyrene. When the concentration of pyrene was 10 mg/L and 50 mg/L, the growth curve of the strain was the same as that of the control group. However, when the concentration of pyrene was 100 mg/L, 150 mg/L and 200 mg/L, the growth of the strain was suppressed but its growth curve was still upward. When the concentration of pyrene was 400 mg/L and 500 mg/L, it could be seen that the growth of the strain was strongly suppressed, but the OD600 value of the bacteria was still increasing slowly (Fig. 8a).

Verification of xenobiotics biodegradation by NY-011. a Growth curve of strain NY-011 in LB medium with different concentrations of pyrene; b Degradation of pyrene by strain NY-011 and the changes of its biomass
Degradation of pyrene by NY-011
During the 28 days, the growth curve of the strain cultured in LB medium added pyrene at 8% NaCl was the same as that without added pyrene. After the strain was transferred into the LB medium with and without added pyrene, respectively, the values of OD600 were 0.256 originally, and then reached the maximum after 7 days, finally decreased slowly. At the same time, after 7 days of culture, the concentration of pyrene in the medium decreased largely, and further decreased with the extension of the culture time, but its trend slowed down, which almost corresponds to the trend of the growth. And after 28 days of culture, the degradation efficiency of pyrene by NY-011 reached up to 36.2% (Fig. 8b).
Discussion
To survive in the high salt conditions, microorganisms require special cell composition and adopt particular adaptation strategies to maintain cellular osmotic balance. To cope with the high salinity environment, halophilic microorganisms have evolved two different strategies to balance the osmotic difference between their cytoplasm and the external environment: (i) the “compatible solutes” strategy, that is to synthesize compatible organic solutes, known as osmotic protectors, in the cells (Edbeib et al. 2016). The types of these small organic molecules used to resist osmotic stress include polyols and their derivatives (such as glycerol), sugars and their derivatives (such as sucrose), amino acids and their derivatives (such as glycine betaine, glutamate, alanine, and ectoine.) and some charged peptides (Galinski 1995); (ii) the “salt in” strategy means completing osmotic balance by accumulating a large number of ions in the cytoplasm from the environment, and the process mainly includes accumulating of K+ and Cl−, expel Na+ (Edbeib et al. 2016). Moderate halophilic bacteria mainly adopt the “compatible solutes” strategy, while extreme halophilic bacteria mainly adopt the “salt in” strategy, but some halophilic microorganisms combine the two strategies (Edbeib et al. 2016; Gunde-Cimerman et al. 2018).
In this study, through genome annotation and transcriptome analysis, we found that NY-011 would simultaneously adopt the two strategies of “compatible solutes” and “salt in” to cope with external salt stress.
The strategy of “compatible solutes” adopted by NY-011 is simultaneously synthesizing alanine, glutamate, ectoine, hydroxyectoine and glycine betaine in cells to maintain osmotic balance. Specifically, NY-011 would firstly synthesize numerous ectoine when exposed to high salt stress, and then convert ectoine to hydroxyectoine with the stress time prolonged. Many studies also showed that the protective effect of hydroxyectoine is more obvious than that of ectoine under more extreme conditions (higher salt concentration or higher temperature) (Argandona et al. 2010; Borges et al. 2002; Van et al. 2013). Besides, results for ectA gene expression obtained by qPCR and by transcriptomics (Fig. 5e) were similar but not identical (the second peak was reached at 15 h for qPCR data and 11 h for transcriptomic data). The reason is the difference between these two methods. Transcriptome sequencing and qPCR are two different detection methods, and a certain degree of inconsistency (about 30 ~ 40%) is normal and reasonable (Su et al. 2014). Factors such as the amplification region of the target gene selected by qPCR, the amplification efficiency of primers, and reagents may affect the final expression level; RNA interruption, library construction, and GC preference in sequencing analysis by RNA-seq may also affect the final results. RNA-seq and qPCR are two different technologies (different principles and different calculation formulas). It is normal for the two to not correspond to each other completely.
In Fig. 5f, glycine betaine synthesis genes (betAB) were up-regulated within 0 ~ 3 h and then their expression dropped, indicating that NY-011 could synthesize a large amount of glycine betaine within 0 ~ 3 h under salt stress. In Fig. 5g, glutamate synthesis gene putA and gdhA were up-regulated in the first hour, and the expression of gdhA dropped at 1 h, putA dropped at 3 h; while glutamate degradation gene glnA and gudB were down-regulated in the first hour, indicating that NY-011 could accumulate a certain amount of glutamate in the cell within 0 ~ 3 h under salt stress. In Fig. 5h, alanine degradation gene agxT2 was down-regulated in the first hour, while alanine synthesis gene alt was up-regulated in the first hour, and alanine synthesis gene ald was up-regulated within 1 ~ 6 h, indicating that NY-011 would synthesize alanine in the initial stage of salt stress. In general, NY-011 mainly synthesizes four kinds of compatible solutes (alanine, glutamic acid, ectoine and glycine betaine) under the short-term osmotic upshock (0 ~ 6 h), while NY-011 mainly synthesizes ectoine and hydroxyectoine under a longer period of osmotic stress. Saum and Müller (2008) have shown that Halobacillus halophilus can synthesize a variety of compatible solutes in cells to cope with high salt stress, and the synthesis of these osmolytes exhibits a time-dependent regulation. They believe that the first response to osmotic stress is the production of chlorine-dependent enzymes that catalyze the biosynthesis of glutamate or glutamine and proline. When the concentration of glutamate or glutamine reaches a certain level, glutamate acting as the second signal produces or activates the enzyme that synthesizes proline and proline is synthesized. Next, the expression of the ect gene may be induced by the third signal. In this process, proline may act as the signal, and then the concentration of proline decreases while the concentration of ectoine increases, and ectoine becomes the main compatible solute in the stationary phase.
The strategy of “salt in” adopted by NY-011 is expelling Na+ from the cell (primary sodium pump and Na+/H+ antiporter) and accumulating K+ in the cell (Trk system and Kdp system). Specifically, the expression of primary sodium pump genes nqrABCD were significantly up-regulated in the first hour and then their expression dropped, while genes of Na+/H+ antiporter were up-regulated within 0 ~ 6 h, and their expression did not reach a significant differential expression level (Fig. 5j). It can be speculated that NY-011 would apply both primary sodium pump and Na+/H+ antiporter to expel Na+ from the cell, and the former seems to play a more important role. The Na+-NQR (Na+-translocating NADH:quinone oxidoreductase) from Vibrio cholerae is a 220 kDa complex consisting of six subunits, NqrABCDEF, encoded by the nqr operon. The Na+-NQR-type enzymes are sodium-motive NADH:quinone oxidoreductases. They can expel sodium ions and maintain low sodium concentration in cells under high salt stress (Fadeeva et al. 2007; Steuber et al. 2014). Under high salt stress, based on transcriptome and proteome data, it was found that Na+-NQR in Halomonas beimenensis was up-regulated expression (Chen et al. 2017).
The genes involved in the K+ transport system were all down-regulated at 1 h, then up-regulated (Fig. 5k), showing that both the Trk system and Kdp system played an important role in accumulating K+ in NY-011. Researches on Escherichia coli show, after an osmotic upshock, the reduced turgor pressure establishes in the cell due to a loss of water. This reduced turgor leads to an influx of K+ within seconds via the potassium transporters (Trk, Kdp and so on). At the same time, glutamate is quickly synthesized in the cell as the main counterion of potassium (Epstein 1986; Stumpe et al. 1996). Potassium glutamate accumulates upon hyper-osmotic shock and serves as a temporary osmoprotectant. This salt leads to transcriptional activation of sets of genes that allow the cell to achieve long-term adaptation to high osmolarity. The current experiments show that potassium glutamate also acts as an inhibitor of bulk cellular transcription (Gralla and Vargas 2006). However, as for why Trk and Kdp systems were down-regulated in the first hour, further research is needed.
Cell morphology of NY-011 under osmotic upshock was observed by SEM and TEM, showing that 15% salt stress will not lead to the lethal damage to NY-011, and it can cope with hypersaline conditions very well. It is worth noting that the cell length of NY-011 increased from 0 to 3 h after the osmotic upshock, and reached the maximum at 3 h. With the extension of stress time, the cell length gradually decreased until at 15 h and returned to the same length as the control group from the results of optical microscope observation and scanning electron microscope observation. Yaakop et al. (2016) also noted that compared to Jeotgalibacillus malaysiensis cells cultured in 2% (w/v) NaCl stress, those cells are grown under 10% and 20% (w/v) NaCl expressed slightly longer. According to our genomics and transcriptomics data (Table S2, Fig. S2), we found that the ftsZ gene in NY-011 was down-regulated, and its expression decreased from 0 to 3 h, reached the minimum at 3 h, and then began to increase until 15 h returned to the same level as that at 0 h. Surprisingly, the expression trend of the ftsZ gene was opposite to the change of cell length. And FtsZ protein was the most important protein in the cell division which can affect cell division (Lutkenhaus et al. 2012). Tao et al. (2017) used CRISPRi technology to inhibit the expression of ftsZ, a gene encoding cell division protein FtsZ in halophilic microorganism Halomonas sp. TD01, and modified the cell morphology to make the bacterial cell length longer. Compared with the control group, the cell length increased by 20 times to 70 times. Therefore, it can be speculated that when the strain was exposed to high salt stress, gene ftsZ was down-regulated and the activity of cell division protein FtsZ decreased, which led to the prolongation of cells and the slowdown of cell growth and reproduction; until the cells adapted to salt stress, with gene ftsZ expressed normally, cells returned to the normal length, and growth and reproduction activities are thriving again.
In addition, we also found PHA-like granules in NY-011 cells by the transmission electron microscope. The genes involved in PHAs synthesis were annotated through genomic data analysis (Table S2), including the key genes phbABC encoding acetyl-CoA acetyltransferase, acetoacetyl-CoA reductase and poly(3-hydroxyalkanoate) synthetase, respectively, which can synthesize PHAs. In the face of hunger or nutritional imbalance, bacteria usually produce PHA granules in the cells as a reserve of carbon source and energy (Anjum et al. 2016). In many prokaryotes, PHAs are accumulated in the form of intracellular granules. In addition to their main function as storage substances, they can also enhance the resistance of bacteria to various external stress. For example, if bacteria are suddenly exposed to osmotic shock, intracellular PHAs can greatly enhance the ability of bacteria to maintain cell integrity (Sedlacek et al. 2019). Furthermore, Obruca et al. (2018) pointed out that PHAs may be accumulated in Cupriavidus necator by applied certain numbers of stress factors, such as osmotic pressure, heavy metals, hydrogen peroxide, which can be employed as an effective and innovative method to boost production of PHAs.
Besides, through GO, COG, and KEGG analysis, a large number of genes related to metabolism, membrane, inorganic ion transport, and degradation of organic pollutants were annotated. Results of RNA-seq showed that when NY-011 was subjected to high salt stress, genes related to osmoregulation were up-regulated at first and then returned to normal expression, while genes related to degradation of organic pollutants were down-regulated and then returned to normal expression. The expression of these two classes of genes was almost the opposite. Thereby, we guess the reason for this phenomenon is that under high salt stress, NY-011 will mobilize all efforts to survive by coping with osmotic stress, so genes related to osmoregulation are up-regulated at first, they are “survival genes”; while other growth and metabolic activities, such as the degradation of organic pollutants, will be weakened for saving the energy, resulting in the down-regulated expression of genes related to degradation of organic compounds, these genes are “growth genes”. With the extension of stress time, NY-011 survived and gradually adapted to osmotic stress, the above-mentioned two classes of genes (“survival genes” and “growth genes”) slowly returned to the normal expression. In other words, we speculate that NY-011 is also able to degrade organic pollutants in the saline environment. To confirm the speculation, the experiment of pyrene degradation by NY-011 at 8% NaCl was conducted. The results showed that after 28 days of culture, the degradation efficiency of pyrene by NY-011 reached up to 36.2%. According to Pernetti and Palma (2005), wastewaters are defined “saline” when the inorganic salt content (generally chlorides and sulphates) is between 0 and 1% w/w; wastewater containing between 1% and 3.5% of salts are defined “highly saline” or “brines”. Therefore, NY-011 can be used to degrade organic pollutants in the high salt environment since 8% NaCl (w/v) [approximately equal to 8% NaCl (w/w)] is far greater than the classification criteria of “highly saline” (3.5% inorganic salt content).
Moreover, in consideration of NY-011 possessing unusual capacities of robustness to stress, degradation of organic pollutants and wide substrate adaptability, it can further be applied in the bioremediation of saline wastewater and soil contaminated by organic compounds. And the optimum conditions (such as temperature, pH, salt concentration etc.) for the maximum biodegradation should be investigated and optimized firstly. Also, to overcome the problems of directly using the pure cultured microorganisms in bioremediation, some effective methods can be utilized, including bioaugmentation, biostimulation, genetic engineering, immobilization technology, bioreactor system and so on.
Abbreviations
- COG:
-
Clusters of Orthologous Groups
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- SEM:
-
Scanning Electron Microscope
- TEM:
-
Transmission Electron Microscope
- PHAs:
-
Polyhydroxyalkanoates
- PAHs:
-
Polycyclic Aromatic Hydrocarbons
- FPKM:
-
Fragments Per Kilobase of transcript sequence per millions of base pairs
- FDR:
-
False Discovery Rate
- NMR:
-
Nuclear Magnetic Resonance
- RT-qPCR:
-
Reverse Transcription Quantitative PCR
- OD600 :
-
Optical Density at 600 nm
- GC-MS:
-
Gas Chromatography-Mass Spectrometry
- SDEGs:
-
Significantly Differentially Expressed Genes
- DEGs:
-
Differentially Expressed Genes
References
Ahmadi M, Jorfi S, Kujlu R, Ghafari S, Darvishi Cheshmeh Soltani R, Jaafarzadeh Haghighifard N (2017) A novel salt-tolerant bacterial consortium for biodegradation of saline and recalcitrant petrochemical wastewater. J Environ Manage 191:198–208
Amini B, Otadi M, Partovinia A (2019) Statistical modeling and optimization of toluidine red biodegradation in a synthetic wastewater using Halomonas strain Gb. J Environ Health Sci Eng 17(1):319–330
Anjum A, Zuber M, Zia KM, Noreen A, Anjum MN, Tabasum S (2016) Microbial production of polyhydroxyalkanoates (PHAs) and its copolymers: a review of recent advancements. Int J Biol Macromol 89:161–174
Argandona M, Nieto JJ, Iglesiasguerra F, Calderon MI, Garciaestepa R, Vargas C (2010) Interplay between iron homeostasis and the osmotic stress response in the halophilic bacterium Chromohalobacter salexigens. Appl Environ Microbiol 76(11):3575–3589
Bairoch A, Apweiler R (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28(1):45–48
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27(2):573–580
Berlin K, Koren S, Chin C-S, Drake JP, Landolin JM, Phillippy AM (2015) Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 33(6):623–630
Besemer J, Borodovsky M (2005) GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33(suppl_2):W451–W454
Borges N, Ramos A, Raven NDH, Sharp RJ, Santos H (2002) Comparative study of the thermostabilizing properties of mannosylglycerate and other compatible solutes on model enzymes. Extremophiles 6(3):209–216
Cao J, Ma H-Y, Li H-Y, Wang K-R, Ruan K, Bai L-H (2013) Halomonas socia sp. nov., isolated from high salt culture of Dunaliella salina. Extremophiles 17(4):663–668
Canovas D, Vargas C, Iglesiasguerra F, Csonka LN, Rhodes D, Ventosa A, Nieto JJ (1997) Isolation and Characterization of Salt-sensitive Mutants of the Moderate Halophile Halomonas elongata and Cloning of the Ectoine Synthesis Genes. J. Biol. Chem. 272(41):25794–25801
Cánovas D, Borges N, Vargas C, Ventosa A, Nieto JJ, Santos H (1999) Role of Ngamma-acetyldiaminobutyrate as an enzyme stabilizer and an intermediate in the biosynthesis of hydroxyectoine. Appl. Environ. Microbiol. 65(9):3774–3779
Ceylan S, Akbulut BS, Denizci AA, Kazan D (2011) Proteomic insight into phenolic adaptation of a moderately halophilic Halomonas sp. strain AAD12. Can J Microbiol 57(4):295–302
Chen Y, Lu C, Shyu Y, Lin S (2017) Revealing the saline adaptation strategies of the halophilic bacterium Halomonas beimenensis through high-throughput omics and transposon mutagenesis approaches. Sci Rep 7(1):13037–13037
Díaz MP, Boyd KG, Grigson SJ, Burgess JG (2002) Biodegradation of crude oil across a wide range of salinities by an extremely halotolerant bacterial consortium MPD-M, immobilized onto polypropylene fibers. Biotechnol Bioeng 79(2):145–153
Delcher AL, Bratke KA, Powers EC, Salzberg SL (2007) Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23(6):673–679
Edbeib MF, Wahab RA, Huyop F (2016) Halophiles: biology, adaptation, and their role in decontamination of hypersaline environments. World J Microbiol Biotechnol 32(8):135
Epstein W (1986) Osmoregulation by potassium transport in Escherichia coli. FEMS Microbiol Lett 39:73–78
Fadeeva MS, Yakovtseva EA, Belevich GA, Bertsova YV, Bogachev AV (2007) Regulation of expression of Na+ -translocating NADH:quinone oxidoreductase genes in Vibrio harveyi and Klebsiella pneumoniae. Arch Microbiol 188(4):341–348
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J (2014) Pfam: the protein families database. Nucleic Acids Res 42(D1):D222–D230
Galinski EA (1995) Osmoadaptation in bacteria. Adv Microb Physiol 37:273–328
Govarthanan M, Khalifa AY, Kamala-Kannan S, Srinivasan P, Selvankumar T, Selvam K, Kim W (2020) Significance of allochthonous brackish water Halomonas sp. on biodegradation of low and high molecular weight polycyclic aromatic hydrocarbons. Chemosphere 243:125389
Gralla JD, Vargas DR (2006) Potassium glutamate as a transcriptional inhibitor during bacterial osmoregulation. The EMBO J 25(7):1515–1521
Gunde-Cimerman N, Plemenitas A, Oren A (2018) Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol Rev 42(3):353–375
Haddadi A, Shavandi M (2013) Biodegradation of phenol in hypersaline conditions by Halomonas sp. strain PH2-2 isolated from saline soil. Int Biodeterior Biodegrad 85:29–34
Jensen LJ, Julien P, Kuhn M, von Mering C, Muller J, Doerks T, Bork P (2007) eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res 36(suppl_1):D250–D254
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28(1):27–30
Kim KH, Jahan SA, Kabir E, Brown RJ (2013) A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects. Environ Int 60:71–80
Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35(9):3100–3108
Lefebvre O, Moletta R (2006) Treatment of organic pollution in industrial saline wastewater: a literature review. Water Res 40(20):3671–3682
Lefebvre O, Vasudevan N, Torrijos M, Thanasekaran K, Moletta R (2005) Halophilic biological treatment of tannery soak liquor in a sequencing batch reactor. Water Res 39(8):1471–1480
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5):589–595
Lutkenhaus J, Pichoff S, Du S (2012) Bacterial cytokinesis: from Z ring to divisome. Cytoskeleton 69(10):778–790
Mccarthy A (2010) Third generation DNA sequencing: pacific biosciences’ single molecule real time technology. Chem Biol 17(7):675–676
Mirbolooki H, Amirnezhad R, Pendashteh AR (2017) Treatment of high saline textile wastewater by activated sludge microorganisms. J Appl Res Technol 15(2):167–172
Mnif S, Chamkha M, Sayadi S (2009) Isolation and characterization of Halomonas sp. strain C2SS100, a hydrocarbon-degrading bacterium under hypersaline conditions. J Appl Microbiol 107(3):785–794
Nzila A, Ramirez CO, Musa MM, Sankara S, Basheer C, Li QX (2018) Pyrene biodegradation and proteomic analysis in Achromobacter xylosoxidans, PY4 strain. Int Biodeterior Biodegrad 130:40–47
Obruca S, Sedlacek P, Koller M, Kucera D, Pernicova I (2018) Involvement of polyhydroxyalkanoates in stress resistance of microbial cells: biotechnological consequences and applications. Biotechnol Adv 36(3):856–870
Oren A, Gurevich P, Azachi M, Henis Y (1992) Microbial degradation of pollutants at high salt concentrations. Biodegradation 3(2–3):387–398
Pernetti M, Palma LD (2005) Experimental evaluation of inhibition effects of saline wastewater on activated sludge. Environ Technol 26(6):695–703
Pradhan S, Fan L, Roddick FA, Shahsavari E, Ball AS (2016) Impact of salinity on organic matter and nitrogen removal from a municipal wastewater RO concentrate using biologically activated carbon coupled with UV/H2O2. Water Res 94:103–110
Saum SH, Müller V (2008) Growth phase-dependent switch in osmolyte strategy in a moderate halophile: ectoine is a minor osmolyte but major stationary phase solute in Halobacillus halophilus. Environ Microbiol 10(3):716–726
Sedlacek P, Slaninova E, Koller M, Nebesarova J, Marova I, Krzyzanek V, Obruca S (2019) PHA granules help bacterial cells to preserve cell integrity when exposed to sudden osmotic imbalances. New Biotech 49:129–136
Steuber J, Halang P, Vorburger T, Steffen W, Vohl G, Fritz G (2014) Central role of the Na(+)-translocating NADH:quinone oxidoreductase [Na(+)-NQR] in sodium bioenergetics of Vibrio cholerae. Biol Chem 395(12):1389–1399
Stumpe S, Schlösser A, Schleyer M, Bakker E (1996) K+circulation across the prokaryotic cell membrane: K+- uptake systems. In: Konings WN, Kaback HR, Lolkema JS (eds) Handbook of biological physics, vol 2. Elsevier, Amsterdam, pp 473–499
Su Z, Łabaj PP, Li S, Thierrymieg J, Thierrymieg D, Shi W, Wang C, Schroth GP, Setterquist R, Thompson JF (2014) A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the sequencing quality control consortium. Nat Biotechnol 32(9):903–914
Tan T, Wu D, Li W, Zheng X, Li W, Shan A (2017) High specific selectivity and membrane-active mechanism of synthetic cationic hybrid antimicrobial peptides based on the peptide FV7. Int J Mol Sci 18(2):339
Tao W, Lv L, Chen G-Q (2017) Engineering Halomonas species TD01 for enhanced polyhydroxyalkanoates synthesis via CRISPRi. Microb Cell Fact 16(1):48
Van TD, Hashim S, Hattikaul R, Mamo G (2013) Ectoine-mediated protection of enzyme from the effect of pH and temperature stress: a study using Bacillus halodurans xylanase as a model. Appl Environ Microbiol 97(14):6271–6278
Yaakop AS, Chan K-G, Ee R, Lim YL, Lee S-K, Manan FA, Goh KM (2016) Characterization of the mechanism of prolonged adaptation to osmotic stress of Jeotgalibacillus malaysiensis via genome and transcriptome sequencing analyses. Sci Rep 6(1):1–14
Zhao Y, Zhuang X, Ahmad S, Sung S, Ni S (2020) Biotreatment of high-salinity wastewater: current methods and future directions. World J Microbiol Biotechnol 36(3):37
Zhang Y, Zhou Y, Sun G, Li K, Li Z, Su A, Liu X, Li G, Jiang R, Han R, Tian Y, Kang X, Yan F (2018) Transcriptome profile in bursa of fabricius reveals potential mode for stress-influenced immune function in chicken stress model. BMC Genomics 19(1):918
Acknowledgements
This work was supported by the Science and Technology Supportive Project of Sichuan Province (2017RZ0063).
Author information
Authors and Affiliations
Contributions
SP participated in the overall design of the experiment, conducted experiments and analyzed data and wrote the manuscript. MK and XY conducted experiments. YL designed physiological experiments and provided experimental conditions. LB designed research and revised the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no financial conflicts of interest.
Ethical statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by M. Moracci.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Peng, S., Kai, M., Yang, X. et al. Study on the osmoregulation of “Halomonas socia” NY-011 and the degradation of organic pollutants in the saline environment. Extremophiles 24, 843–861 (2020). https://doi.org/10.1007/s00792-020-01199-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-020-01199-5