
Abstract
In order to assess bacterial diversity within four surface sediment samples (0–5 cm) collected from the Pacific Arctic Ocean, 16S ribosomal DNA clone library analysis was performed. Near full length 16S rDNA sequences were obtained for 463 clones from four libraries and 13 distinct major lineages of Bacteria were identified (α, β, γ, δ and ε-Proteobacteria, Acidobacteria, Bacteroidetes, Chloroflexi, Actinobacteria, Firmicutes, Planctomycetes, Spirochetes, and Verrucomicrobia). α, γ, and δ-Proteobacteria, Acidobacteria, Bacteroidetes, Actinobacteria were common phylogenetic groups from all the sediments. The γ-Proteobacteria were the dominant bacterial lineage, representing near or over 50% of the clones. Over 35% of γ-Proteobacteria clones of four clone library were closely related to cultured bacterial isolates with similarity values ranging from 94 to 100%. The community composition was different among sampling sites, which potentially was related to geochemical differences.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Benthic bacterial communities in the ocean environment play a significant role in the global biogeochemical cycle, because they can rapidly degrade and utilize particulate organic matter (Gooday and Turley 1990; Kostka et al. 1999; Rysgaard et al. 1999; D’Hondt et al. 2002; Bowman et al. 2003). Studies have shown that the fraction of bacteria in the deep sub-seafloor biosphere may make up one-tenth to one-third of the Earth’s total biomass and about 70% of the global prokaryotic biomass (Parkes et al. 1994; Whitman et al. 1998). As a result, bacterial communities are not only functioning in degradation, but they are also an important component in the food web structure.
Microbial community structure analysis is important for an understanding of benthic ecosystem processes and in defining the roles that benthic bacteria play in overall oceanic processes. Prior studies have revealed that a complex microbial community is buried within marine sediment, although most sequences were distantly related to cultured bacteria (Gray and Herwig 1996; Li et al. 1999a, b; Teske et al. 2002; Zeng et al. 2005; Polymenakou et al. 2005; Webster et al. 2006). Molecular analysis of coastal polar sediments also indicated the presence of a rich, uncultivated bacterial diversity in sediments (Ravenschlag et al. 1999, 2001; Llobet-Brossa et al. 1998; Bowman and McCuaig 2003). A number of studies on bacterial community in the Atlantic Arctic marine sediments had been carried out, especially in the area around Svalbard (Sahm and Berninger 1998; Sahm et al. 1999; Ravenschlag et al. 1999, 2000, 2001; Knoblauch et al. 1999). The focus of these studies was mostly specific microbial groups such as sulfate-reducing bacteria in this habitat. An intensive clone library data from cold marine sediment collected at Hornsund off the coast of Spitsbergen indicated a predominance of sequences related to bacteria of the sulfur cycle, and the δ and γ-Proteobacteria were dominated in the sediment (Ravenschlag et al. 1999). The Cytophaga-Flavobacterium cluster, along with the γ-Proteobacteria and sulfate reducers, was one of the three most abundant groups in the top 5 cm of Svalbard fjord sediments by fluorescent in situ hybridization (FISH) and rRNA hybridization (Ravenschlag et al. 2001). Numerous γ and δ Proteobacterial phylotypes dominated the community and community composition changed significantly between the thin oxic layer and the anoxic zone below, which was reported from an Antarctic continental shelf surficial sediment clone library analysis. Many sediment bacterial phylotype groups were widespread and always present in marine sediments, as shown by an extensive comparison of available clone library data (Bowman and McCuaig 2003).
The Pacific Arctic Region is loosely defined as the area lying between Russia and Alaska (Bering Strait), extending northward including the Beaufort Gyre and Arctic Ocean and south including the Bering Sea, and also including seasonally frozen regions (http://www.pagscience.org/aboutus.html). Research in the Beaufort Gyre and Canada Basin had been extremely limited due to seasonal or the heavy year-round ice cover. Knowledge of the biodiversity and community structure of benthic bacteria in this area is still lacking. However, scientific research in this area received new impetus during the last decade. Several expeditions focused on exploring the deep Canada Basin, located in the Arctic Ocean (Gradinger and Bluhm 2005), including two expeditions carried out by Chinese groups in 1999 and 2003 (Chen 2000; Zhang 2004). Our study presents 16S rDNA clone library of four surface sediments collected from the Chukchi Sea and the deep Canada Basin in order to obtain a preliminary understanding of bacterial community composition in this region.
Materials and methods
Sample collection and DNA extraction
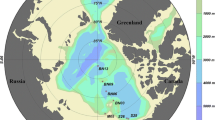
Four surface sediment samples (0–5 cm) were taken from undisturbed box cores (35 × 35 × 65 cm) during the 2nd Chinese National Arctic Research Expedition between 11 and 28 August in 2003 (Fig. 1). Within each box core, the top 5-cm samples were taken from the center and four corners using a 5-cm diameter tube and carefully mixed well. Station S11 and S26 located on the Chukchi shelf slope with depths of 40 and 3,000 m, respectively. Station P13 located on the Chukchi Plateau at the depth of 447 m. Station B78 was within the Canada Basin at a depth of 3,850 m (Table 1). Samples were kept frozen during transportation and stored at −20°C until used. Total community genomic DNA was extracted from 1 or 10 g wet weight sediment samples using an Ultra clean Soil DNA kit (Mo Bio Laboratories, Carlsbad, CA) following the manufacturer’s protocol.

Station locations for four sediments during the CHINARE 2003 studies
16S rDNA clone library construction
PCR amplification of 16S rDNA was performed with an Eppendorf Mastercycler Gradient (Eppendorf, Germany) and the bacterial primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) (Bosshard et al. 2000). In a final volume of 50 μl, the PCR reaction mixture contained: 1.0 μl of template DNA, 5.0 μl of 10× PCR buffer (Sangon, Shanghai, China), 40 μM dNTPs, 0.2 μM of each described primers and 1U Taq DNA polymerase (Sangon, Shanghai, China). The PCR reaction started with pre-denaturation at 95°C for 4 min followed by 25 cycles of denaturation at 95°C for 1 min, annealing at 50°C for 1 min and extension at 72°C for 2 min, with a final extension at 72°C for 10 min.
Clone library construction, screening and processing followed Webster et al. (2006). Briefly, each gene library was constructed from four independent PCR products, which were pooled and purified with the gel extraction kit (Watson, Shanghai, China) according to the manufacturer’s instructions. Cloning was conducted with pGEM-T Vector (Promega) following manufacturer’s instructions. Libraries were screened for the 1.5 kb 16S rDNA insert by PCR with M13 primers. Full-length inserts were sequenced with M13 primers on an ABI PRISM 3730 sequencer.
Phylogenetic analysis
Ribosomal RNA gene sequences from clone libraries were checked for chimeras with Chimera Check from the RDP II (Maidak et al. 2001) and their closest relatives identified by NCBI BLAST (http://www.ncbi.nlm.nih.gov/). All nucleotide sequences were aligned using Clustal X1.8 (Thompson et al. 1997) with nearly complete sequences retrieved from the database. Alignments were edited manually using BioEdit Sequence Alignment Editor version 5.0.9 (Hall 1999) and regions of ambiguous alignment were removed. Phylogenetic trees were constructed using neighbor-joining with the Kimura 2-parameter correction algorithm in MEGA version 4 (Tamura et al. 2007). A series of 1,000 bootstrap data sets of the same size as the original nucleotide sequence data were re-sampled using the SEQBOOT option of the MEGA software.
Operational taxonomic unit richness estimation
In a community, the species richness increases with the sampling effort. The relationship between the species richness and sampling effort can be used to estimate the total richness of a community from a sample; and the estimates can then be compared among samples (Hughes et al. 2001). To conduct richness estimation and rigorous comparison among the communities, sequences were placed into operational taxonomic units (OTUs) at a level of sequence similarity of ≥97%. All OTU richness and sample coverage calculations were performed with the program EstimateS (version 8.0, http://viceroy.eeb.uconn.edu/estimates). For the purposes of importing the data into the program, each cloned sequence was treated as a separate sample, and 100 randomizations were conducted for all the tests. Further randomizations did not change the results. The OTU richness was calculated for each of the sediment samples using the non-parametric estimator Chao 1 (Chao 1987). Extrapolation using best-fit regression analysis was performed (where necessary) to calculate the point at which 95% confidence intervals (CIs) did not overlap (Hughes et al. 2001).
Nucleotide sequence accession numbers
The almost full-length 16S rDNA sequences determined in this study were deposited in the GenBank database under Accession Nos. EU286965–EU287427.
Results
Analysis of bacterial 16S rDNA clone libraries
In total, 214, 174, 213 and 223 bacterial 16S rDNA clones were PCR screened, and 144, 107, 128, 149 positive transformants were obtained from the B78, P13, S11 and S26 libraries, respectively. After sequencing and chimera checking, a total of 463 clones of nearly full-length 16S rDNA sequences from the four bacterial clone libraries (129 from B78, 90 from P13, 117 from S11 and 126 from S26) were submitted to further analysis. The majority of sequences were similar to 16S rDNA sequences in GenBank with similarities ranging from 90 to 100%. Sequences having ≥97% similarity were assigned to the same phylotype, 64, 77, 53 and 72 phylotypes were defined within B78, P13, S11 and S26 libraries, respectively. The relative distribution of the major phylogenetic groups within each library is shown in Fig. 2, the relative richness among libraries is shown in Fig. 3, and the phylogenetic relationships among clones are displayed in Figs. 4, 5, 6, and 7.

Distribution of bacterial 16S rRNA gene sequences from the Pacific Arctic Ocean

OTU estimate curves derived from 16S rDNA clone library data

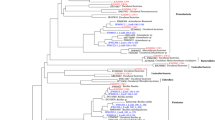
Phylogenetic relationships of bacterial 16S rDNA sequences within γ-Proteobacteria from the Pacific Arctic Ocean clone libraries that related to uncultured clones. The trees were inferred by neighbor-joining analysis. Bootstrap support values over 50% (1,000 replicates) are shown. Bold type indicates clones in this study, and the numbers in parentheses are the number of closely related sequences with 94–100% similarity in the same library. Scale bar indicates the estimated number of base changes per nucleotide sequence position

Phylogenetic relationships of bacterial 16S rDNA sequences within γ-Proteobacteria from the Pacific Arctic Ocean clone libraries that related to cultured isolates. The trees were inferred by neighbor-joining analysis. Bootstrap support values over 50% (1,000 replicates) are shown. Bold type indicates clones in this study, and the numbers in parentheses are the number of closely related sequences with 94–100% similarity in the same library. Scale bar indicates the estimated number of base changes per nucleotide sequence position

Phylogenetic relationships of bacterial 16S rDNA sequences within α, β-Proteobacteria from the Pacific Arctic Ocean clone libraries. The trees were inferred by neighbor-joining analysis. Bootstrap support values over 50% (1,000 replicates) are shown. Bold type indicates clones in this study, and the numbers in parentheses are the number of closely related sequences with 94–100% similarity in the same library. Scale bar indicates the estimated number of base changes per nucleotide sequence position

Phylogenetic relationships of bacterial 16S rDNA sequences within δ, ε-Proteobacteria, Bacteroidetes, Acidobacteria, Actinobacteria, Spirochetes, Chloroflexi, Planctomycetes, Verrucomicrobia and Fimicutes from the Pacific Arctic Ocean clone libraries. The trees were inferred by neighbor-joining analysis. Bootstrap support values over 50% (1,000 replicates) are shown. Bold type indicates clones in this study, and the numbers in parentheses are the number of closely related sequences with 94–100% similarity in the same library. Scale bar indicates the estimated number of base changes per nucleotide sequence position
In this study, although diverse bacterial lineages were detected, some sequences were common in all four sediments (Figs. 4, 5, 6, 7). The most abundant clones in our libraries were affiliated with Proteobacteria (including α, β, γ, δ and ε-Proteobacteria), especially γ-Proteobacteria. The remaining phylotypes appeared to be scattered over a broad range of taxons, e.g., Acidobacteria, Bacteroidetes, Chloroflexi, Actinobacteria, Firmicutes, Planctomycetes, Spirochetes, and Verrucomicrobia. Moreover, most of the clones from the libraries were similar to environmental sequences recovered from Arctic or Antarctic marine sediments, sea ice, seawater and lake water. Since these clusters did not contain cultivated prokaryotes, they were designated arbitrarily with names of corresponding 16S rDNA clones derived from earlier studies. Almost all diversity studies on the deep marine biosphere use the term Green non-sulfur bacteria (GNS) for the Chloroflexi (Garrity et al. 2002), and GNS was used here to facilitate comparisons with previous studies. Based on the criterion that 93% similarity corresponds to a taxonomic grouping at the genus levels (Mullins et al. 1995), sequences with similarities of 94–100% to known sequences were defined as ‘closely related’ in this study.
γ-Proteobacteria
As shown in Fig. 2, γ-Proteobacteria were the most commonly sampled group present within the studied sediment, representing 41, 59, 76 and 61% of clones within B78, P13, S11 and S26 libraries, respectively. A large proportion of γ-Proteobacterial phylotypes detected, however, were grouped into 10 clusters distinct from cultured species (Fig. 4). These groups included only clones detected previously in ice-covered Antarctic lake sediment, marine sediment samples (from both coastal and deep-sea sites) and Arctic sea-ice samples. The largest group (ELB16-053), as a unique representative phylotype in S11 library (65 related clones, 63 clones from S11 library), branched deeply within the γ-Proteobacteria and was associated with the clone ELB16-053 from Antarctic Lake Bonney water (Glatz et al. 2006). Cluster ELB19-048 (8 related clones), ELB16-074 (7 related clones) within γ-Proteobacteria were a minor fraction, similar to sequences from the same Antarctic lake. Five clusters (JT58-28, JTB255, BD3-6, D6 and BD1-1) grouped with the deep-sea bacterial clones detected in previous studies. The second cluster JT58-28 (18 related clones within S26 library) was distantly related to (91% similarity) the Methylobacter clone JT58-28 of cold-seep sediments from Japan Trench (GenBank AB189348). The third cluster (15 clones in P13 and S11 libraries) was affiliated with the bacterial clones JTB255 from cold seep sediments from the Japan Trench (Li et al. 1999b). The sequences within cluster BD3-6 (13 clones) and BD1-1 (7 clones) were very similar to the 16S rDNA sequences of clone from deep-sea sediments (Li et al. 1999a). The cluster D6 was related to the single clone recovered from the sediment sample from Pacific warm pool with high similarity (96–100%) (Zeng et al. 2005). The sequences of Belgica2005/10-140-11 and ARKIA-103 formed distinct clusters that were closely related to the clone sequences of a metal contaminated coastal sediment (Gillan and Pernet 2007), and Arctic pack ice (Brinkmeyer et al. 2003). With the exception of S11 and P13 libraries, there was little overlap between B78 and S26 libraries among the γ-Proteobacteria related to uncultured clones (Fig. 4). For example, the cluster JTB255, ELB19-048 and BD1-1 included the most of sequences detected in S11 and P13 libraries. Overall, the JT58-28 and ELB16-074 sequences were special bacterial phylotypes in the S26 library, whereas clones grouped within D6 and ARKIA-103 appeared to be a feature in B78 library.
About one quarter (103 out of 463 clones within the four libraries, containing 35% γ-Proteobacteria) of the sequences within the γ-Proteobacteria were similar to cultured chemoheterotrophic genera such as Colwellia, Alcanivorax, Shewanella, Marinobacter, Aeromonas, Aranicola, Glaciecola, Legionella, Pseudomonas, Alteromonas and Marinmonas (Fig. 5), most of them occupied cold marine ecosystems (sea ice, sea water or sediment). The most significant proportion (34 clones of S26 library) was closely related to Colwellia sp. cultivated from Arctic seawater by our group (GenBank EF551379). In B78 library, 21 clones were similar to Colwellia psychroerythrus IC064 isolated from Antarctic sea ice (Bowman et al. 1997). Thirteen sequences recovered from sediment B78, P13 and S11 belonged to genus Alcanivorax, associated with an isolate cultivated from the Arctic sediment sampled from the same area (GenBank DQ514306). Six clone sequences of S11 library and two of B78 library were closely related to a halophilic bacterium Marinobacter sedimentalis R65T, which was isolated from Peter the Great Bay sediment sample, Sea of Japan, Russia (Romanenko et al. 2005) and a Marinobacter strain cultivated by our group from Arctic sea-ice sample (GenBank DQ514303). Seven related clones from the S26 library were closely related to Shewanella baltica OS155 (GenBank CP000563), as was a single clone in the P13 library. Other genera (Aeromonas, Aranicola, Glaciecola, Legionella, Pseudomonas, Alteromonas and Marinmonas) were represented by a small number of sequences retrieved from B78, P13 and S26 libraries, and related to cultured isolates from marine environments. For example, two sequences in the B78 library grouped with Glaciecola pallidula ACAM 615T (Bowman et al. 1997). Two clone sequences of P13 library were 88% similar to Legionella sp. OA32, an intracellular bacterium of marine dinoflagellate (GenBank AB058916). Clone S26-93, S26-46 and P13-15 were closely related to Aeromonas molluscorum 431E isolated from bivalve molluscs (Minana-Galbis et al. 2002, 2004).
α and β-Proteobacteria
The α and β-Proteobacteria groups were shown together in Fig. 6. The β-Proteobacteria sequences made up about 40% of the B78 library and were practically absent in the S11 library (Fig. 2). Within this group, abundant sequences of B78 fell into two main distinct clusters (ZD0412 and WLB13-215), and were closely related to the sequences detected in Antarctic lake water (Glatz et al. 2006) and North Sea (Zubkov et al. 2002), respectively. Only a few clones similar to cultivated isolates. Besides clone B78-86 was closely related to a phoxin-degrading bacterium ZM-1 of genus Delftia (GenBank EF061135), clone P13-38 and S26-9 were similar to an ammonia-oxidizing isolate Nitrosospira sp. III7 from a spruce forest, Norway (Purkhold et al. 2003).
In the S26 library, 17% of clones fell within the α-Proteobacteria, but the abundance decreased to 6, 9 and 1% in the P13, B78 and S11 libraries. The majority of clones within the α-Proteobacteria formed two main clusters (Rhodobacteraceae and Kordiimonas). In the Rhodobacteraceae clade, besides two clones closely affiliated with the isolate 20188, a Phaeobacter sp. recovered from Arctic sediment samples by our group (GenBank DQ514304), other clones were similar to Roseobacter denitrificans OCh 114 from oligotrophic marine environment (Swingley et al. 2007), or Sulfitobacter sp. clone F4C15 of sub-Antarctic seawater (Prabagaran et al. 2007), or Antarctic lake water clone ELB16-059 (Glatz et al. 2006). Cluster Kordiimonas contained the clones closely related to a bacterium Kordiimonas gwangyangensi GW14-5 isolated from marine sediments (Kwon et al. 2005). Other minor clusters were grouped with clones of uncultured species recovered from deep-sea sediments (BD1-8, Li et al. 1999a) or associated with a gutless worm (Blazejak et al. 2006) and sponge (Sorensen et al. 2007), all from marine habitats.
δ-Proteobacteria and Acidobacteria
In this study, most clone sequences of the δ-Proteobacteria were detected in the S11 (12%) and P13 (10%) libraries (Fig. 2) and formed four clusters (Fig. 7). Three clusters (Sva0081, Sva0566 and BD1-2) of δ-Proteobacteria were obtained in S11 library. The cluster Sva0081 and Sva0566 grouped with bacterial clone Sva0081 and Sva0566, respectively, found in Arctic permanently cold marine sediments, which was affiliated with Desulfosarcina sp. and Desulfuromonas sp (Ravenschlag et al. 1999). The other cluster was related to the deep-sea clone BD1-2 (Li et al. 1999a). Most of the δ-Proteobacterial clones in P13 library formed one cluster, which was allied with a bacterial clone HMMVProg-8 obtained from the marine sediments, Barents sea (Losekann et al. 2007).
Clones grouped within Acidobacteria of P13 library accounted for 14% (Figs. 2, 7), the most significant of which similar to the clone sequence VHS-B5-22 found in the harbor sediments, Victoria Harbour, Hong Kong (Zhang et al. 2008). A few clones from the P13, B78 and S26 libraries branched with ocean crust clone P0X3b1D02 detected in seafloor lavas from Hawai’i South Point X3 (Santelli et al. 2008).
Bacteroidetes, Actinobacteria and others
Sequences within Bacteroidetes group comprised 15% of S26 library, while they made up only a small portion of S11, B78 and P13 libraries (Fig. 2). In this study, about 50% of Bacteroidetes clones were most similar to cultured members of the genera Flexibacter, Zobellia, Flavobacterium, Maribacter (Nedashkovskaya et al. 2007), most of which were isolated from deep-sea sediments.
In all libraries, sequences belonging to Actinobacteria group were revealed in small proportions (Fig. 2). This group formed two clusters associated with clone BD7-2 and Sva0996 (Fig. 7), respectively. Five clone sequences of the B78 library were related to the uncultured clone Sva0996 presented in Arctic cold sediment (Ravenschlag et al. 1999) and clones from deep-sea sediment of Pacific nodule province (GenBank AJ966592 and AM162470). The other minor cluster was similar to the bacterial clone BD7-2 of the deep-sea sediment at a depth of 6,379 m (Li et al. 1999a).
The ε-Proteobacteria, Chlorofexi, Firmicutes, Spirochetes, Verrucomicrobia and Planctomycetes contained a small number of clone sequences (Fig. 2), most of which were closely related to uncultured clones derived from marine environmental samples (Fig. 7). The clone sequences within ε-Proteobacteria and Spirochetes only presented in S11 library, which were grouped with the Guaymas Basin hydrothermal vent sediments clone B01R002 allied to the sulfate-reducing bacteria (Dhillon et al. 2003) and clone IE094 described in GenBank. Sequences of Verrucomicrobia and Planctomycetes were found exclusively in the P13 library and closely related to the clone recovered from coastal sediments on the Belgian continental plate (Gillan and Pernet 2007) and cold-seep sediments in Japan Trench (GenBank AB189369). One clone from S26 library and two clones from B78 library grouped with the Chloroflexus-related clone PK016 retrieved from Bahamas marine sponge associated microbial (Taylor et al. 2007).
OTUs richness estimation of four sediment clone library
As the clone libraries were constructed at the same time, the bacterial diversity of Pacific Arctic Ocean sediments clone libraries were subjected to comparative analysis to extrapolate species richness. Plotting the cumulative number of OTUs estimated against the sampling effort gives species richness curves (Fig. 3). The highest estimated number of species was evidently present in the P13 sediment sample, which contained 475 OTUs (95% CIs, 201 and 1,377). whereas the B78, S11 and the S26 sediments samples yielded 225 OTUs (95% CIs, 88 and 774), 193 OTUs (95% CIs, 105 and 430) and 317 OTUs (95% CIs, 160 and 737), respectively (Fig. 3). The 95% CIs for the B78, P13, S26 and S11 sediments communities overlapped, and thus at the P level of 0.05 there was no significant difference in species richness among the samples.
Discussion
This study provides characterization of the four surface sediments collected from the Pacific Arctic Ocean by 16S rDNA libraries. Overall, a total of 463 bacterial 16S rDNA clones from the four sediment samples revealed rich phylogenetic diversity (Figs. 4, 5, 6, 7). All sequences derived from the four sediment samples fell into thirteen major bacterial phylogenetic lineages including α, β, γ, δ and ε-Proteobacteria, Acidobacteria, Bacteroidetes, Chloroflexi, Actinobacteria, Firmicutes, Planctomycetes, Spirochetes, and Verrucomicrobia. Moreover, six major bacterial lineages (α, γ, δ-Proteobacteria, Bacteroidetes, Actinobacteria, Acidobacteria) were detected in the four sediments. Molecular studies of the bacterial communities of other cold saline sediment such as the High Arctic cold saline spring sediments (Perreault et al. 2007), Antarctic continental shelf (Bowman and McCuaig 2003), Japan Trench deep-sea cold seep (Li et al. 1999b) and Shikoku Basin surface sediments (Mu et al. 2005) also revealed highly diverse bacterial populations. Many of the common phylotypes were represented by identical sequences between our clones and the sequences previously detected in other marine surface, deep-sea cold seeps and cold saline spring sediments. The Proteobacteria were the most cosmopolitan group, occurring very frequently in Pacific Arctic sediments and in all other marine sediments (Table 2). The Proteobacteria are also the dominant bacterial phylogenetic lineage in most surface marine sediment, comprising >50% of the microbial biomass (Bowman et al. 2003; Ravenschlag et al. 2001).
There was clear trends in the dominant bacterial lineages found in Pacific Arctic Ocean surface sediments. In all sediments, the γ-Proteobacteria of Bacteria was predominated. It seems that the γ-Proteobacteria were the most significant clades in most marine sediments (Li et al. 1999a, b; Bowman and McCuaig 2003; Inagaki et al. 2003; Zeng et al. 2005; Polymenakou et al. 2005). For example, when seven deep-sea sediment samples taken from different depths (1,159–6,482 m) were investigated, the results show that the sequences within the γ-Proteobacteria were dominant in all of the sediments studied (Li et al. 1999a). Besides the δ-Proteobacteria sulfate reducer, the γ-Proteobacteria sulfur oxidizer phylotypes were found to dominate in Arctic Hornsund fjord marine sediments (Ravenschlag et al. 1999). The γ-Proteobacteria also dominated in volcanic ash layer of deep sediments sampled from the Sea of Okhotsk (Inagaki et al. 2003). As detected by FISH and real-time PCR (Ravenschlag et al. 2001; Bowman et al. 2005), γ-Proteobacteria was a significant part of the bacterial community in polar marine sediment samples. In the upper 2 cm layers, γ-Proteobacteria accounted for up to 10.5% of the total cell counts, and 20% of prokaryotic rRNA, in the Smeerenburgfjorden sediments (Ravenschlag et al. 2001). Bowman et al. (2005) quantified the abundant uncultured γ-Proteobacterial marine sediment (GMS) clades from Antarctic continental shelf sediment samples using SYBR Green-based real-time PCR, and they found that GMS clades made up 0.3–8.7% of total 16S rRNA genes.
In our four libraries, a fraction of sequences within the γ-Proteobacteria with high sequence similarity to cultured bacteria (such as genera Colwellia, Shewanella, Alcanivorax and Pseudomonas) was detected (Fig. 5). This result was identical well with previous studies, although different culturable genera were found in different marine sediments. In deep-sea sediments, the most common sequences were Pseudomonas-related sequences (Li et al. 1999a). The clones in the γ-Proteobacteria from the surface layer of Antarctic Mertz cores were closely related to cultivated bacterial genera Shewanella, Pseudoalteromonas and Colwellia (Bowman and McCuaig 2003), whereas the γ-Proteobacteria clones from Sea of Okhotsk sediments were grouped with cultured bacterial genera Halomonas, Colwellia, Shewanella, Mehylophaga and Psychrobacter (Inagaki et al. 2003). It is possible that the γ-Proteobacteria comprise chemoheterotrophic bacteria, which dominate benthic sediment environments. FISH analysis of the enrichments from German North Sea water showed a complex community that was dominated by the γ-Proteobacteria (Uphoff et al. 2001). It was suggested that the versatile γ-Proteobacteria can grow rapidly on a wide range of carbon sources of different concentrations, leading to their dominance in rich growth media.
However, besides the dominant groups, the presence and percentages of other bacterial phylogenetic clusters in the four clone libraries were divergent (Fig. 2, Table 2). Overall, bacterial phylogenetic groups of sediment samples collected from the shallow and deeper water overlapped little in this study(Figs. 4, 5, 6, 7), likely reflecting the differences of the pressure between the sampling sites. Significantly, a large portion of clone sequences (39% of total clones) from the sediment B78 clustered into the β-Proteobacteria, whereas they were nearly absent in the S11 library. Considering the β-Proteobacteria are characteristically found in freshwater habitats (Methé et al. 1998), this suggests that the terrestrial influence on the sediment B78 was greater than other three sediments. One of the most important features of the Arctic Ocean is its terrestrial context, being surrounded by continents and receiving very significant freshwater influxes (Bano and Hollibaugh 2002). Total organic carbon is primarily delivered to the Arctic Ocean via major rivers such as the Mackenzie in Canada and the Yenisei, Lena, and Ob in Eurasia. The annual sediment load to the Arctic from the Mackenzie River alone was estimated at 127 Mt (Macdonald et al. 1998). But the characteristics of sediments in the Arctic Ocean basins was closely correlated to many environmental factors including terrestrial inputs from rivers, water depth, distance from the shore, circulation, topographical features and sea-ice transport (Zhang 2004). It was found that though total organic matter on the shallow Chukchi Shelf was higher than those in the Canada Basin, surface sediment of this area was dominated by substantial autochthonous marine carbon due to high primary production of micro-alga and significant degradation before the organic matter reaches the sediment (Belicka et al. 2002). Moreover they found that the Transpolar Drift and the Lomonosov Ridge appeared to influence the transport and focusing of terrestrial material in the Arctic Ocean basins. Thus the observed differences in bacterial community composition among these sediments were likely to be associated with their geographic location. Further study should be undertaken to understand the relationship between bacterial community composition and geochemical characteristics of the different location in the Pacific Arctic in the future.
References
Bano N, Hollibaugh JT (2002) Phylogenetic composition of bacterioplankton assemblages from the Arctic Ocean. Appl Environ Microbiol 68:505–518
Belicka LL, Macdonald RW, Harvey HR (2002) Sources and transport of organic carbon to shelf, slope, and basin surface sediments of the Arctic Ocean. Deep-Sea Res I 49:1463–1483
Blazejak A, Kuever J, Erseus C, Amann R, Dubilier N (2006) Phylogeny of 16S rRNA, ribulose 1, 5-bisphosphate carboxylase/oxygenase, and adenosine 5′-phosphosulfate reductase genes from gamma- and alphaproteobacterial symbionts in gutless marine worms (oligochaeta) from Bermuda and the Bahamas. Appl Environ Microbiol 72:5527–5536
Bosshard PP, Santini Y, Grüter D, Stettler R, Bachofen R (2000) Bacterial diversity and community composition in the chemocline of the meromictic alpine lake Cadagno a reveal by 16S rDNA analysis. FEMS Microbiol Ecol 31:173–182
Bowman JP, McCuaig RD (2003) Biodiversity, community structural shifts, and biogeography of prokaryotes within Antarctic continental shelf sediment. Appl Environ Microbiol 69:2463–2483
Bowman JP, McCammon SA, Brown MV, Nichols DS, McMeekin TA (1997) Diversity and association of psychrophilic bacteria in Antarctic sea ice. Appl Environ Microbiol 63:3068–3078
Bowman JP, McCammon SA, Gibson JAE, Robertson L, Nichols PD (2003) Prokaryotic metabolic activity and community structure in Antarctic continental shelf sediment. Appl Environ Microbiol 69:2448–2462
Bowman JP, McCammon SA, Dann AL (2005) Biogeographic and quantitative analyses of abundant uncultivated γ-Proteobacterial clades from marine sediment. Microb Ecol 49:451–460
Brinkmeyer R, Knittel K, Jurgens J, Weyland H, Amann R, Helmke E (2003) Diversity and structure of bacterial communities in Arctic versus Antarctic pack ice. Appl Environ Microbiol 69:6610–6619
Chao A (1987) Estimating the population size for capture recapture data with unequal catchability. Biometrics 43:783–791
Chen L (2000) The report of the first Chinese Arctic Research Expedition (in Chinese). China Ocean Press, Beijing
Dhillon A, Teske A, Dillon J, Stahl DA, Sogin ML (2003) Molecular characterization of sulfate-reducing bacteria in the Guaymas Basin. Appl Environ Microbiol 69:2765–2772
D’Hondt S, Rutherford S, Spivack AJ (2002) Metabolic activity of subsurface life in deep-sea sediments. Science 295:2067–2070
Garrity GM, Winters M, Kuo AW, Searles DB (2002) Taxonomic outline of the Prokaryotes, Bergey’s Manual of Systematic Bacteriology, 2nd edn edn. Springer, New York
Gillan DC, Pernet P (2007) Adherent bacteria in heavy metal contaminated marine sediments. Biofouling 23:1–13
Glatz RE, Lepp PW, Ward BB, Francis CA (2006) Planktonic microbial community composition across steep physical/chemical gradients in permanently ice-covered Lake Bonney, Antarctica. Geobiology 4:53–67
Gooday AJ, Turley CM (1990) Responses by benthic organisms to inputs of organic material to the ocean floor: a review. Phil Trans R Soc Lond A 331:119–138
Gradinger R, Bluhm BA (2005) Arctic Ocean exploration, 2002. Polar Biol 28:169–170
Gray JP, Herwig RP (1996) Phylogenetic analysis of the bacterial communities in marine sediments. Appl Environ Microbiol 62:4049–4059
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser No.41 41:95–98
Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJM (2001) Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol 67:4399–4406
Inagaki F, Suzuki M, Takai K, Oida H, Sakamoto T, Aoki K, Neaslon KH, Horikoshi K (2003) Microbial communities associated with geological horizons in coastal subseafloor sediments from the Sea of Okhotsk. Appl Environ Microbiol 69:7224–7235
Knoblauch C, Jørgensen BB, Harder J (1999) Community size and metabolic rates of psychrophilic sulfate-reducing bacteria in Arctic marine sediments. Appl Environ Microbiol 65:4230–4233
Kostka JE, Thamdrup B, Glud RN, Canfield DE (1999) Rates and pathways of carbon oxidation in permanently cold Arctic sediments. Mar Ecol Prog Ser 180:7–21
Kwon KK, Lee HS, Yang SH, Kim SJ (2005) Kordiimonas gwangyangensis gen. nov., sp. nov., a marine bacterium isolated from marine sediments that forms a distinct phyletic lineage (Kordiimonadales ord. nov.) in the ‘Alphaproteobacteria’. Int J Syst Evol Microbiol 55(PT 5):2033–2037
Li L, Kato C, Horikoshi K (1999a) Bacterial diversity in deep-sea sediments from different depths. Biodivers Conserv 8:659–677
Li L, Kato C, Horikoshi K (1999b) Microbial diversity in sediments collected from the deepest cold-seep area, the Japan Trench. Mar Biotechnol 1:391–400
Llobet-Brossa E, Rossello-Mora R, Amann R (1998) Microbial community composition of Wadden Sea sediments as revealed by fluorescence in situ hybridization. Appl Environ Microbiol 64:2691–2696
Losekann T, Knittel K, Nadalig T, Fuchs B, Niemann H, Boetius A, Amann R (2007) Diversity and abundance of aerobic and anaerobic methane oxidizers at the Haakon Mosby mud volcano, Barents Sea. Appl Environ Microbiol 73:3348–3362
Macdonald RW, Solomon SM, Cranston RE, Welch HE, Yunker MB, Gobeil C (1998) A sediment and organic carbon budget for the Canadian Beaufort Shelf. Marine Geol 144:255–273
Maidak BL, Cole JR, Lilburn TG, Parker CT, Saxman PR, Farris RJ, Garrity GM, Olsen GJ, Schmidt TM, Tiedje JM (2001) The RDP-II (Ribosomal Database Project). Nucleic Acids Res 29:173–174
Methé BA, Hiorns WD, Zehr JP (1998) Contrasts between marine and freshwater bacterial community composition: analyses of communities in Lake George and six other Adirondack lakes. Limnol Oceanogr 43:368–374
Minana-Galbis D, Farfan M, Loren JG, Fuste MC (2002) Biochemical identification and numerical taxonomy of Aeromonas spp. isolated from environmental and clinical samples in Spain. J Appl Microbiol 93:420–430
Minana-Galbis D, Farfan M, Fuste MC, Loren JG (2004) Aeromonas molluscorum sp. nov. isolated from bivalve mollusks. Int J Syst Evol Microbiol 54:2073–2078
Mu C, Bao Z, Chen G, Hu J, Hao L, Qi Z, Li G (2005) Bacterial diversity in the sediments collected from the Shikoku Basin. Acta Oceanol Sinica 24:114–121
Mullins TD, Britschgi TB, Krest RL, Giovannoni SJ (1995) Genetic comparisons revealed that the same unknown bacterial lineages in Atlantic and Pacific bacterioplankton communities. Limno Oceanogr 40:148–158
Nedashkovskaya OI, Vancanneyt M, De Vos P, Kim SB, Le MS, Mikhailov VV (2007) Maribacter polysiphoniae sp. nov., isolated from a red alga. Int J Syst Evol Microbiol 57(PT 12):2840–2843
Prabagaran SR, Manorama R, Delille D, Shivaji S (2007) Predominance of Roseobacter, Sulfitobacter, Glaciecola and Psychrobacter in seawater collected off Ushuaia, Argentina, Sub-Antarctica. FEMS Microbiol Ecol 59(2):342–355
Parkes RJ, Cragg BA, Bale SJ, Getliff JM, Goodman K, Rochelle PA, Fry JC, Weightman AJ, Harvey SM (1994) Deep bacterial biosphere in Pacific Ocean sediments. Nature 371:410–413
Perreault NN, Andersen DT, Pollard WH, Greer CW, Whyte LG (2007) Characterization of the prokaryotic diversity in cold saline perennial springs of the Canadian high Arctic. Appl Environ Microbiol 73:1532–1543
Polymenakou PN, Bertilsson S, Tselepides A, Stephanou EG (2005) Bacterial community composition in different sediments from the eastern Mediterranean Sea: a comparison of four 16S ribosomal DNA clone libraries. Microb Ecol 50:447–462
Purkhold U, Wagner M, Timmermann G, Pommerening-Roser A, Koops HP (2003) 16S rRNA and amoA-based phylogeny of 12 novel betaproteobacterial ammonia-oxidizing isolates: extension of the dataset and proposal of a new lineage within the nitrosomonads. Int J Syst Evol Microbiol 53(Pt 5):1485–1494
Ravenschlag K, Sahm K, Pernthaler J, Amann R (1999) High bacterial diversity in permanently cold marine sediments. Appl Environ Microbiol 65:3982–3989
Ravenschlag K, Sahm K, Knoblauch C, Jørgensen BB, Amann R (2000) Community structure, cellular rRNA content and activity of sulfate reducing bacteria in marine arctic sediments. Appl Environ Microbiol 66:3592–3602
Ravenschlag K, Sahm K, Amann R (2001) Quantitative molecular analysis of the microbial community in marine arctic sediments (Svalbard). Appl Environ Microbiol 67:387–395
Romanenko LA, Schumann P, Rohde M, Zhukova NV, Mikhailov VV, Stackebrandt E (2005) Marinobacter bryozoorum sp. nov. and Marinobacter sediminum sp. nov., novel bacteria from the marine environment. Int J Syst Evol Microbiol 55:143–148
Rysgaard S, Thamdrup B, Risgaard-Petersen N, Fossing H, Berg P, Christensen PB, Dalsgaard T (1999) Seasonal carbon and nutrient mineralization in a high-Arctic coastal marine sediment, Young Sound, Northeast Greenland. Mar Ecol Prog Ser 175:261–276
Sahm K, Berninger UG (1998) Abundance, vertical distribution and community structure of benthic prokaryotes from permanently cold marine sediments (Svalbard, Arctic Ocean). Mar Ecol Prog Ser 165:71–80
Sahm K, MacGregor BJ, Jørgensen BB, Stahl DA (1999) Sulphate reduction and vertical distribution of sulphate-reducing bacteria quantified by rRNA slot-blot hybridization in a coastal marine sediment. Environ Microbiol 1:65–74
Santelli CM, Orcutt BN, Banning E, Bach W, Moyer CL, Sogin ML, Staudigel H, Edwards KJ (2008) Abundance and diversity of microbial life in ocean crust. Nature 453(7195):653–656
Sorensen KB, Glazer B, Hannides A, Gaidos E (2007) Spatial structure of the microbial community in sandy carbonate sediment. Mar Ecol Prog Ser 346:61–74
Swingley WD, Sadekar S, Mastrian SD, Matthies HJ, Hao J, Ramos H, Acharya CR, Conrad AL, Taylor HL, Dejesa LC, Shah MK, O’huallachain ME, Lince MT, Blankenship RE, Beatty JT, Touchman JW (2007) The complete genome sequence of Roseobacter denitrificans reveals a mixotrophic rather than photosynthetic metabolism. J Bacteriol 189(3):683–690
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Taylor MW, Radax R, Steger D, Wagner M (2007) Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol Mol Biol Rev 71(2):295–347
Teske A, Hinrichs KU, Edgcomb V, de Vera Gomez A, Kysela D, Sylva SP, Sogin ML, Jannasch HW (2002) Microbial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities. Appl Environ Microbiol 68:1994–2007
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Uphoff HU, Felske A, Fehr W, Wagner-Dobler I (2001) The microbial diversity in picoplankton enrichment cultures: a molecular screening of marine isolates. FEMS Microbiol Ecol 35:249–258
Webster G, Parkes RJ, Fry JC, Weightman AJ (2004) Widespread occurrence of a novel division of bacteria identified by 16S rRNA gene sequences originally found in deep marine sediments. Appl Environ Microbiol 70:5708–5713
Webster G, Parkes RJ, Cragg BA, Newberry CJ, Weightman AJ, Fry JC (2006) Prokaryotic community composition and biogeochemical processes in deep subseafoor sediments from the Peru Margin. FEMS Microbiol Ecol 58:65–85
Whitman WB, Coleman DC, Wiebe WJ (1998) Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 95:6578–6583
Zhang Z (2004) The report of 2003 Chinese Arctic Research Expedition (in Chinese). Ocean press, Beijing
Zhang W, Ki JS, Qian PY (2008) Microbial diversity in polluted harbor sediments I: Bacterial community assessment based on four clone libraries of 16S rDNA. Estuar Coast Shelf Sci 76(3):668–681
Zeng R, Zhao J, Zhang R, Lin N (2005) Bacterial community in sediment from the western Pacific Warm Pool and its relationship to environment. China Environ. Sci 48:282–290
Zubkov MV, Fuchs BM, Archer SD, Kiene RP, Amann R, Burkill PH (2002) A population of the alpha-proteobacteria dominates the bacterioplankton and dimethylsulphoniopropionate uptake after an algal bloom in the North Sea. Deep-sea Res II, Top Stud Oceanogr 49(15):3017–3038
Acknowledgments
We appreciate the assistance of the crew of the Xuelong and the scientists who collected samples for us on the CHINARE 2003 cruise, especially A.G. Gao, R.J. Wang and M.H. Cai. H.B. Teng, Q. Luo and M. Wang provided useful data analysis and other support during the preparation of the manuscript. Y.H. Su and A.B. Chen helped with sample analysis. We also thank Dr. H.S. Bi for assistance in editing and discussing this manuscript. This work was supported by the National Natural Science Foundation of China (No. 40876097, 40806073), Chinese National Basic Research Program (No. 2004CB719601), Polar Strategic Research Foundation of China, and the program of the Science and Technology Committee of Shanghai, China (052307053). This work is also a part of the Project ‘Second Chinese National Arctic Research Expedition’ or CHINARE-2003 supported by the Ministry of Finance of China and organized by the Chinese Arctic and Antarctic Administration (CAA). The participants in this joint work are from the institutions (e.g., PRIC, FIO, SIO, etc.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Robb.
Rights and permissions
About this article
Cite this article
Li, H., Yu, Y., Luo, W. et al. Bacterial diversity in surface sediments from the Pacific Arctic Ocean. Extremophiles 13, 233–246 (2009). https://doi.org/10.1007/s00792-009-0225-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-009-0225-7