
Abstract
A high-alkaline, salt-activated alginate lyase is produced by Agarivorans sp. JAM-A1m from a deep-sea sediment off Cape Nomamisaki on Kyushu Island, Japan. Purified to homogeneity, as judged by SDS-PAGE, the enzyme (A1m) had a molecular mass of approximately 31 kDa. The optimal pH was around 10 in glycine–NaOH buffer, and the activity was increased to 1.8 times by adding 0.2 M NaCl. However, when the optimal pH in the presence of 0.2 M NaCl was shifted to pH 9.0, the activity was more than 10 times compared with that at pH 9 in the absence of NaCl. A1m showed the optimal temperature at around 30°C and was stable to incubation between pH 6 and 9. The enzyme degraded favorably mannuronate–guluronate and guluronate-rich fragments in alginate. Shotgun cloning and sequencing of the gene for A1m revealed a 930-bp open reading frame, which encoded a mature enzyme of 289 amino acids (32,295 Da) belonging to polysaccharide lyase family 7. The deduced amino acid sequence showed the highest similarity to that of a Klebsiella enzyme, with only 54% identity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
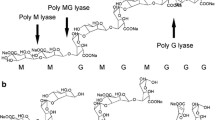
Alginate is a linear hetero-polyuronic acid consisting of 1,4 linked α-l-guluronic acid (G) and its C5 epimer, β-d-mannuronic acid (M). These residues are arranged in block structures that comprise a homopolymeric G block, M block, alternating MG (GM) block, and heteropolymeric MG (GM) blocks (Gacesa 1988). Alginates are synthesized as a cell wall component by brown seaweeds and also as an exopolysaccharide by bacteria such Pseudomonas aeruginosa (Evans and Linker 1973) and Azotobacter vinelandii (Gorin and Spencer 1966).
Unlike pectate or pectin decomposing enzymes, alginate-degrading enzymes are known to be only lyases (transeliminase) even if they are intracellular oligo-alginate-degrading enzymes. Alginate lyses have been isolated from various sources, namely, marine algae, marine mollusks, fungi, bacteria, bacteriophages, and viruses (Wong et al. 2000). They are classified into three groups by their substrate specificity toward G block (polyguluronate lyase; EC4.2.2.11), M block (polymannuronate lyase; EC4.2.2.3), or MG block (Wong et al. 2000). Furthermore, they are also grouped into three types based on their molecular masses: small (25–30 kDa), medium-sized (around 40 kDa), and large lyases (>60 kDa) (Osawa et al. 2005). Thus, it is easily supposed that alginate lyases show a wide molecular diversity. Indeed, alginate lyases are classified into seven polysaccharide lyase (PL) families (http://www.cazy.org/fam/acc_PL.html). Many alginate lyases have optimal pH values between pH 7 and 8, in contrast to the optimal high-alkaline pH values of pectate lyases (Wong et al. 2000). Alkaline alginate lyases are very few, and the most alkaline enzyme reported to date is derived from the Chlorella virus, CVN1 (Suda et al. 1999) with an optimal pH of 10.5; however, there is no report of its substrate specificity. Other lyases with highly alkaline optimal pH of 9.2–9.6 that degrade the M block as a substrate have been isolated from the invertebrates, Aplysia depilans (Boyen et al. 1990b) and Turbo cornutus (Muramatsu et al. 1984). Among alginate lyase-producing bacteria, only one alkaliphilic Bacillus sp. No. M-2 has been reported to produce an alkaline enzyme, with an optimal pH of 9 (Horikoshi and Akiba 1982). The enzyme is salt-activated and degrades the M block in alginate. This means that isolation of a highly alkaline G or MG block-degrading enzyme from a microorganism has never been reported. Recently, we found a new highly alkaline alginate lyase (A1m) from a deep-sea bacterium, Agarivorans sp. JAM-A1m. In this article, we describe screening and purification of the enzyme and cloning and sequencing of the gene for the enzyme.
Materials and methods
Isolation of alginate lyase-producing bacteria
Deep-sea sediments were collected off Cape Nomamisaki, at the southwestern tip of Kyushu Island (representative sampling site: 31°18.522N, 129°59.372E), Japan, at a depth of 254 m in July 2005. Aliquots of the samples were spread onto marine agar 2216 (Difco) containing 0.2% sodium alginate (from Laminaria, 80–120 cps; Wako Pure Chemicals). After cells had grown at 30°C for 24 h, a 0.8% agar solution containing 0.2% sodium alginate and 50 mM MOPS buffer (pH 7) was poured over the agar. After incubation at 30°C for 24 h, 0.1% cetylpyridinium chloride solution was flowed onto the soft agar. Colonies that formed clear zones were picked up and purified several times on marine agar 2216. The isolates were propagated aerobically in large test tubes containing 5 ml of marine broth 2216 (Difco) plus 0.2% sodium alginate at 30°C for 24 h. Alginate lyase activities in the supernatants were measured in 100 mM acetate buffer (pH 5), MOPS buffer (pH 7), or glycine–NaOH buffer (pH 9) at 30°C for 1 h. The alginate lyases of four isolates among ten candidates showed higher activity in glycine–NaOH buffer (pH 9). One of the isolates, strain JAM-A1m, showed the highest productivity, and the enzyme showed the highest activity at pH 10 in glycine–NaOH buffer.
Determination of 16S rRNA gene sequence of isolate
To clarify the phylogenetic position of strain A1m, the 16S rRNA gene sequence was determined by colony PCR. The gene was amplified using bacterial primers 27f (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492r (5′-GGCTACCTTGTTACGACTT-3′), and a small pieces of strain A1m as the template in a DNA thermal cycler (Gene Amp PCR system 9700, ABI PRISM) with an LA Taq DNA polymerase. PCR conditions were 2 min at 96°C, followed by 30 cycles of 30 s at 96°C, 30 s at 55°C, and 90 s at 72°C, and final extension for 7 min at 72°C. After purification of the amplified gene fragment, sequencing was done with a DNA sequencer (Mega BACE 1000, GE Healthcare) using a DYEnamic ET Terminal Sequencing kit (GE Healthcare). The nucleotide sequence of the 16S rRNA gene has been submitted to GenBank/EMBL/DDBJ databases under accession no AB426125. Strain A1m has been deposited at the National Institute of Advanced Industrial Science and Technology (AIST) under deposit number FERM P-21397.
Production and purification of A1m
Agarivorans sp. JAM-A1m was propagated aerobically, with shaking, in 100 ml of medium optimized for production of A1m at 30°C for 24 h. The medium was composed of (w/v) 0.5% polypeptone S (Nihon Pharmaceutical), 0.5% yeast extract (Difco), 2% NaCl, 0.2% sodium alginate, 0.02% MgSO4·7H2O, 0.01% K2HPO4, and 0.002% MnCl2. Sodium alginate was indispensable for the production of A1m, because the enzyme was inducible. The spent media were centrifuged (8,000×g for 20 min at 4°C), and the supernatant (2.5 l) was concentrated to 290 ml using a hollow fiber filter (AIP0013, 6 kDa cut off, Asahikasei). The concentrate was applied to a SuperQ Toyopearl column (2.5 × 8 cm; Tosoh) equilibrated with 50 mM Tris–HCl buffer (pH 8.0). The absorbed proteins were eluted with a 450-ml linear gradient of 0–0.22 M NaCl in the equilibrating buffer. Alginate lyase activity was eluted around 0.16 M NaCl. Ammonium sulfate was added to the pooled fraction (75 ml) to a final concentration of 2 M. The solution was applied to a Butyl Toyopearl column (1.5 × 5 cm; Tosoh) equilibrated with 10 mM Tris–HCl buffer (pH 8.0) containing 2 M ammonium sulfate. Proteins were eluted with a 120-ml linear gradient of 2–0.6 M ammonium sulfate in 10 mM Tris–HCl buffer. The active fractions were obtained at around 1.4 M ammonium sulfate. The active fractions were combined, and the same procedures were repeated except that a linear gradient of 2–0.5 M ammonium sulfate was used. The active fractions were concentrated and desalted by ultrafiltration using a YM3 membrane (Millipore).
Determination of molecular mass of A1m
Molecular mass of the purified enzyme was determined by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and gel permeation chromatography. SDS-PAGE was performed by the method of Laemmli (1970) using a 12.5% acrylamide slab gel (Bio-Rad). The proteins were stained with Coomassie Brilliant Blue R-250. Precision plus protein standards (Bio-Rad) were used as the molecular mass markers. Bio-Gel-A0.5 m gel column (1.5 × 75 cm; Bio-Rad) was equilibrated with 0.2 M KCl in 10 mM Tris–HCl buffer (pH 7.5). The standard proteins used were conalbumin (75 kDa), ovalbumin (43 kDa), carbonic anhydrase (30 kDa), and lysozyme (14.2 kDa).
Analysis of N-terminal amino acid sequence
After SDS-PAGE, the protein band was electrotransferred onto a polyvinylidene difluoride membrane (Imobulon; Millipore) that had been wetted with methanol. The amino acid sequences were determined by a protein sequencer (model 497HT; Applied Biosystems).
Enzyme assay and protein content
Alginate lyase activity was measured in a 0.5 ml of reaction mixture composed of 0.2% (w/v) sodium alginate, 100 mM glycine–NaOH buffer (pH 10), and a suitably diluted enzyme solution. After incubation at 30°C for 15 min, the reaction was terminated by adding 2 ml of 5 mM HCl, and then the absorbance of the solution was measured at 235 nm. One unit of enzyme activity was defined as the amount of protein that increased absorbance by 0.1 per min at 235 nm. To confirm the products of the enzymatic reaction, thiobarbituric acid (TBA) test was used according to Weissbach and Hurwitz (1959). The viscometric assay was done using as Ostwald viscometer (No.1; Shibata Scientific Technology). The reaction mixture included 0.2% sodium alginate, 100 mM glycine–NaOH buffer (pH 9), 200 mM NaCl, and a suitably diluted enzyme solution in a total volume of 10 ml. The decrease of viscosity in the mixture was measured at 30°C at timed intervals. The absorbance at 235 nm (A 235) of timed sample (0.5 ml) was also measured after adding 2 ml of 5 mM HCl. The protein content was quantified by a DC protein assay kit (Bio-Rad) using bovine serum albumin as the standard.
Preparation of substrates
Mannuronic acid (M) and guluronic acid (G) blocks were prepared by oxalic acid hydrolysis of sodium alginate basically according to the method of Haug et al. (1966). M and G blocks were qualitatively confirmed by infrared spectroscopy (type JIR-7000; JEDR). For the G block, the absorptions around 1,415, 1,324, 1,124, 1,091, 1,027, and 949 cm−1 showed greater intensity, whereas the intensities of the M block around 1,144, 1,039, and 820 cm−1 were clearer and larger than those for G block (Sartori et al. 1997; Pereira et al. 2003). MG blocks were prepared from partial acid hydrolysis of 5% (w/v) sodium alginate in 0.3 N HCl at 100°C for 20 min as described by Haug et al. (1967).
Cloning and sequencing gene for A1m
The genomic DNA of Agarivorans sp. JAM-A1m, which was prepared by the method of Saito and Miura (1963), was digested with Sau3AI. DNA fragments (0.75–3 kb) were ligated into BamHI site of pUC18. Escherichia coli DH5α was transformed by the resultant plasmids. After transformants were grown on Luria-Bertani agar containing 0.1% sodium alginate plus 100 μg ml−1 ampicillin, a soft agar solution (0.2% alginate, 0.2 M NaCl, 50 mM glycine–NaOH buffer, and 0.8% agar, pH 9) was poured over the agar plate. The agar was incubated at 30°C for 24 h, and then 0.1% cetylpyridinium chloride solution was poured onto the soft agar. Transformants showing alginate-degrading zones were picked up and purified several times on LB agar containing 100 μg ml−1 ampicillin. Several plasmids were isolated from the cultured transformants by a High Pure Plasmid Isolation kit (Roche), and subsequently inserted DNA fragments were amplified by PCR using primers A, 5′-CAAGGCGATTAAGTTGGGTAACG-3′ and B, 5′-CTTCCGGCTCGTATGTTGTGTG-3′, which were designed from the outside of multiple cloning sites in the pUC18 plasmid. The amplified fragments were ligated into a pCR2.1 using a TA Cloning kit (Invitrogen) and were transformed into E. coli TOP10. After blue/white colony selection of transformants, cloned DNA fragments were amplified by colony PCR using primers A and B and were sequenced using a DYEnamic ET Terminal Sequencing kit and a DNA sequencer. The A1m gene was also sequenced and confirmed by PCR using primers that were designed from the above sequence and the genomic DNA of Agarivorans sp. JAM-A1m as the template. Nucleotide sequence data of A1m gene have been submitted to the GenBank, EMBL, and DDBJ databases under accession no AB426616.
Results
Analysis of 16S rRNA gene sequence of strain JAM-A1m
A 1,497-bp fragment of the 16S rRNA gene of strain JAM-A1m was sequenced and compared with those of other strains. The highest nucleotide sequence match was that of Agarivorans albus MKT87 (AB076559) with 98.6% identity (1,453 of 1,473 bases match). The next best match was observed on that of Agarivorans sp. JAMB-A11 (AB167508) with 98.4% identity (1,411 of 1,434 bases match). These results indicate that strain JAM-A1m is closely related to the genus Agarivorans.
Purification and physicochemical properties of A1m
A1m was purified to homogeneity as judged by SDS-PAGE from spent media of Agarivorans sp. JAM-A1m. A typical purification was summarized in Table 1. The purification increased the specific activity 764 times with 6% recovery of the initial activity. The molecular mass of the enzyme was around 31 kDa by SDS-PAGE (Fig. 1). A1m was eluted at the same position of carbonic anhydrase by Bio-Gel-A0.5m gel filtration column chromatography, indicating that the enzyme is monomer. The N-terminal amino acid sequence of the purified A1m was Ala-Thr-Thr-Thr-Pro-Ala-Glu-Val-Leu-Asp-Leu-Ser-X-X-X-Val-Thr, where X is an unidentified amino acid residue.

SDS-PAGE of purified A1m. Lane A protein mass markers; lane B purified enzyme (5 μg). The proteins were subjected to electrophoresis on 12.5% acrylamide gel and stained with Coomassie Brilliant Blue R-250
Effects of pH and NaCl on A1m
The optimal pH of A1m activity was around pH 10 in 100 mM glycine–NaOH buffer. Very low activity was observed between pH 6 and 11 except around pH 10. When 0.2 M NaCl was added to the reaction mixture, the activity was increased to 1.8 times the original activity, and then the optimal pH was shifted to pH 9 in glycine–NaOH buffer. The activity was increased to more than 10 times that at pH 9 in the absence of NaCl. Similar results were observed in 100 mM borate buffer with or without 0.2 M NaCl, as shown in Fig. 2. In contrast, the stability of A1m in various pH buffers was unaffected by adding NaCl. A1m was quite stable in incubation at 30°C for 1 h between pH 6 and 9 (Fig. 3). Although the residual activities were only 60% of those in the absence of NaCl, the enzyme was modestly stable between pH 6 and 7 under the same conditions in the presence of 0.2 M NaCl.

Effect of pH on A1m activity. The activity was measured at 30°C for 15 min in a total volume of 0.5 ml containing 2.2 or 1.1 μg of purified A1m in the absence (solid line) or presence of 0.2 M NaCl (dotted line), respectively. The following 100 mM buffers were used: open triangles MOPS (pH 6–7.9); filled circles Tris–HCl (pH 7.1–8.9); open circles borate–NaOH (pH 9–10.3); filled triangles glycine–NaOH (pH 8–10.8). The highest activity, obtained at pH 9 in glycine–NaOH buffer plus 0.2 M NaCl, is taken as 100%

Effect of pH on A1m stability. Purified A1m (1 μg) was preincubated at 30°C for 1 h in various 20 mM buffers with (dotted line) or without (solid line) 0.2 M NaCl. After preincubation, the residual activity was measured at 30°C for 15 min in 100 mM glycine–NaOH buffer (pH 9) plus 0.2 M NaCl. The buffers used were acetate (pH 4–6, filled squares), MOPS (pH 6–8, open triangles), Tris–HCl (pH 7–9, filled circles), glycine–NaOH (pH 9–11, filled triangles), and phosphate–NaOH (pH 11–12.3, open squares). The highest residual activity treated in 20 mM Tris–HCl (pH 7) is taken as 100%
Effects of NaCl on A1m
The effect of the NaCl concentration on the enzyme activity was examined. A1m activity at pH 9 in glycine–NaOH buffer increased with increasing NaCl concentration, and the activity reached maximum at 0.6–0.8 M NaCl, which was around 20 times without NaCl (Fig. 4). Although the effect of NaCl on activation of A1m activity at pH 10 was less than that at pH 9, the activity increased within the broad concentration of 0.1–0.6 M NaCl to approximately double that without NaCl (Fig. 4). Thus, A1m is one of the most salt-activated enzymes among alginate lyases reported to date (Horikoshi and Akiba 1982; Kitamikado et al. 1992; Wong et al. 2000; Xiao et al. 2006). The kinetic parameters for alginate degradation in the presence or absence of NaCl at pH 9 were determined by double reciprocal Lineweaver Burk plots. The apparent Km values were 0.032% regardless of whether NaCl was present or not. The apparent V max values were 38.4, 285.7, 416.7, and 526.3 units mg−1 protein in the presence of 0, 0.1, 0.2, and 0.5 M NaCl, respectively. These results suggest that the catalytic efficiency of Alm for alginate degradation is increased by adding NaCl.

Effect of NaCl concentration on A1m activity. The activity was measured at 30°C for 15 min in 100 mM glycine–NaOH buffer with the indicated NaCl concentrations. The purified enzyme (2.7 or 1.1 μg) was used in a reaction mixture at pH 10 (filled circles with solid line) or at pH 9 (open circles with dotted line). The activity at pH 9 in glycine–NaOH buffer plus 0.2 M NaCl is taken as 100%
Effects of temperature of A1m
The optimal temperature for the reaction of A1m was around 30°C either in the presence of 0.2 M NaCl at pH 9 or in its absence at pH 10. Activation of the enzyme by NaCl was prominent between 25 and 35°C (Fig. 5). As expected, A1m is quite thermolabile. A1m is rapidly inactivated upon incubation at 30–50°C for 15 min in 20 mM glycine–NaOH buffer (pH 9) with or without 0.2 M NaCl. The residual activity after treatment at 30°C with 0.2 M NaCl was only 53% of the residual activity without NaCl. However, relative inactivation rates were similar whether NaCl was present or not. The residual activities after treatment at 50°C were only 10% of the initial activities after incubation at 30°C with or without NaCl.

Effect of temperature on A1m activity. Alginate lyase activity was measured in 100 mM glycine–NaOH buffer at the indicated temperatures for 15 min with 2.2 or 1.1 μg of purified enzyme in the absence (pH 10, filled circles with solid line) or presence of 0.2 M NaCl (pH 9, open circles with dotted line), respectively. The maximal activity in the presence of 0.2 M NaCl is taken as 100%
Effects of other salts and metal ions on A1m activity
A1m was activated by several salts, including 0.2 M each NaF, NaBr, NH4Cl, (NH4)2SO4, KCl, and K2HPO4 (Table 2). Pd2+, Hg2+, and Sr2+ (1 mM each) ions inhibited the enzyme by 95, 80, and 46%, respectively. Ca2+ and Mg2+ ions did not affect enzyme activity unlike other alginate lyases (Wong et al. 2000; Svanem et al. 2001; Xiao et al. 2006). NaCl was the best activator among tested salts and metal ions. The enzyme was strongly inhibited by 5 mM EDTA and EGTA (Table 2). Dithiothreitol and 2-mercaptoethanol (5 mM each) did not affect the enzyme activity at all.
Substrate specificity
A1m preferably degraded the MG and G blocks to the M block. The relative activities for alginate, MG, G, and M blocks were 100, 131.7 ± 16.5, 83.3 ± 16.9, and 27.3 ± 13.4%, respectively, under the standard assay conditions at pH 10. The mean value and standard deviation were calculated from four experimental results. Similar results were observed when measured at pH 9 with 0.2 M NaCl.
The viscosity of an alginate solution rapidly decreased by adding A1m, whereas the values of A 235 increased very slowly (Fig. 6), suggesting that the enzyme degrades alginate by endo fashion. The products of the enzymatic reaction were positive for TBA method. This means that 4-deoxy-l-erythro-hex-4-enopyranosyluronic acids as the non-reducing terminal moiety are contained in the products. In our assay, it showed good correlation between the values of A 235 per mg protein and those of TBA test (A550 per mg protein) with a ratio of 0.25 (A 235/A 550).

Viscosity reduction during enzymatic degradation of alginate. A1m (1.5 μg) was added to the reaction mixture composed of 100 mM glycine–NaOH buffer (pH 9), 0.2% sodium alginate and 0.2 M NaCl in a final volume of 10 ml and, at intervals, viscosity in the solution was measured at 30°C. The initial viscosity of the reaction mixture without enzyme was taken as 100%. At timed intervals, samples (0.5 ml) were also withdrawn from the paranemic reaction mixture, and 2 ml of 5 mM HCl was added, followed by measurement of absorbance at 235 nm. Open circles with solid line rate of viscosity reduction; filled circles with dotted line absorbance at 235 nm
Nucleotide and deduced amino acid sequences for A1m
The shotgun-cloned DNA fragments contained one open reading frame of 930 bp. It started from an ATG codon and terminated at a TAG codon. The G+C content of ORF was 42.6%. A putative ribosome binding site, with the sequence 5′-TATAGG-3′, was found 8 bp upstream of the initiation codon. An inverted repeat sequence was found 20 bp downstream of the termination codon. The free energy value of the stem-loop structure was calculated to be −69.1 kJ mol−1. The ORF encoded 309 amino acids including a putative signal sequence of 20 amino acids, as shown in Fig. 7. In the signal sequence of Met1 to Ala20, several basic amino acid residues after Met1, and a helix-broken Gly12 were found, and Leu18-Ser19-Ala20 was supposed to be a signal peptidase recognition sequence. Indeed, the N-terminal amino acid sequence of the purified A1m from Agarivorans sp. JAM-A1m was found to be Ala21 to Thr37 in the deduced amino acid sequence. Thus, the mature enzyme consists of Ala21 to Lys309, with a calculated molecular mass of 32,295 Da, a value that is close to the 31 kDa of the purified A1m. When suitably aligned, the deduced amino acid sequence of A1m exhibited some homology to those of known alginate lyases belonging to polysaccharide lyase family 7. A1m showed the highest similarity to AlyA (Q059478) of Klebsiella pneumonia subsp. aerogenes with 53.7% identity. It showed moderate similarity to putative alginate lyases of Vibrio splendidus 12B01 (A3UR41), Saccharophagus degradans strain 2–40 (Q21HU1), Plesiocystis pacifica SIR-1 (A6G2Q0), and Polaribacter dokdonensis MED152 (A2TZT7) with 50.7, 49.7, 49.1, and 48.1% identities, respectively, which are merely annotated by genome analysis. These results suggest that A1m is a new alginate lyase.

Nucleotide and deduced amino acid sequences of A1m. Deduced amino acid residues are represented by single letters under the nucleotide sequence. The N-terminal amino acid sequence determined from the purified enzyme is underlined. The amino acid sequences that are highly conserved among G-block-degrading enzymes in PL family 7 are boxed, and identical amino acid residues are shown in bold letters
Discussion
There have been many reports of alginate lyases from various origins, such as algae, marine animals, marine bacteria, marine fungi, viruses, and other gram-positive and gram-negative bacteria (Wong et al. 2000; Suzuki et al. 2006; Kawamoto et al. 2006; Iwamoto et al. 2001). In marine bacteria, many alginate lyases have been isolated from members of Pseudomonas and Vibrio (Wong et al. 2000). This study is the first report that the genus Agarivorans from a deep-sea sediment produced an alginate lyase. The Agarivorans enzyme (A1m) favorably degraded the MG- and G-blocks in alginate. An alginate lyase from Pseudomonas sp. (Min et al. 1977), and AlyVG1, AlyVG2 and AlyVG3 from Vibrio halioticoli (Sugimura et al. 2000) also show the similar degradation specificity. The optimal pH of A1m was around pH 10 in both glycine and borate buffers, and it shifted to pH 9 when NaCl was added to the reaction mixture. This is the highest optimal pH reported for alginate lyases from microorganisms. In particular, the value is the highest among MG or G block-degrading enzymes. Interestingly, A1m was remarkably activated by salts, probably the cationic ions, Na+, NH4 +, and K+ ions. Although salt-activated G block-degrading lyases, such as the alginate lyases AL-1 and AL-2 of marine bacterium ALG-G (Brown and Preston 1991), and the alginate lyases of Klebsiella aerogenes type 25 (Lange et al. 1989), Pseudomonas alginovora strain Xo17 (Boyen et al. 1990a), and Vibrio harveyi AL-128 (Kitamikado et al. 1992), are known, none of them shifted their optimal pH value when activated by adding salts. Furthermore, A1m was not activated by Ca2+ and/or Mg2+ ions unlike the G block-specific degrading alginate lyases from P. alginovora strain Xo17 (Boyen et al. 1990a) and Corynebacterium sp. ALY-1 (Matsubara et al. 1998). The optimal temperature of A1m was around 30°C, a relatively low optimal temperature among known alginate lyases (Wong et al. 2000). A1m was quite stable in incubation at 30°C for 1 h between pH 6 and 9, whereas the enzyme was relatively unstable under the same conditions in the presence of 0.2 M NaCl. The reason why the enzyme was unstable in the presence of NaCl is not clear.
Alginate lyases are organized into polysaccharide lyase (PL) families 5, 6, 7, 14, 15, 17, and 18, which show wide diversity compared with other polysaccharide lyases. Recently, the crystal structures of several alginate lyases belonging to PL family 7 have been resolved and the active site and hydrogen network clarified (Osawa et al. 2005; Yamasaki et al. 2004, 2005). Among them, the structures of PL family 7 alginate lyases, namely the G block- degrading ALY-1 of Corynebacterium sp. (Osawa et al. 2005) and A1-II′ of Sphingomonas sp. A1 (Yamasaki et al. 2005), and the M block-degrading PA1167 of Pseudomonas aeruginosa (Yamasaki et al. 2004), show the jellyroll β-sandwich fold with a large active cleft. According to their structures and sequences analyses, amino acid residues in active sites of PL family 7 enzymes are well conserved depending on their substrate specificity (see Fig. 7).
A1m is a highly alkaline enzyme compared with the other PL family 7 enzymes. We are interested in alkaline adaptation of proteins. We have proposed three alkaline adaptation-strategies of enzymes. The first strategy aims at shifting the pKa of the catalytic site residues toward alkaline pH. The second one aims at acquiring the higher ratio of surface-exposed Asp + Glu to Lys + Arg for the alkaline enzymes. The last one aims at decreasing Lys and Asp residues and at increasing Arg, His, and Glu residues in the alkaline-adaptation process (Shirai et al. 2008). The Asp + Glu/Lys + Arg (40/33) of A1m is larger than that (33/34) of AlyA of K. aerogenes which shows the highest similarity to A1m but is a neutrophilic enzyme (Wong et al. 2000). Although the crystal structures of A1m and AlyA are not resolved, this result is in accordance with the second strategy of alkaline adaptation. A determination of the crystal structure of A1m will be helpful to further clarify the alkaline adaptation mechanism.
References
Boyen C, Bertheau Y, Barbeyron T, Kloareg B (1990a) Preparation of guluronate lyase from Pseudomonas alginovora for protoplast isolation in Laminaria. Enzyme Microb Technol 12:885–890
Boyen C, Kloareg B, Polne-Fuller M, Gibor A (1990b) Preparation of alginate lyases from marine molluscs for protoplast isolation in brown algae. Phycologia 29:173–181
Brown BJ, Preston JF (1991) l-guluronan-specific alginate lyase from a marine bacterium associated with Sargassum. Carbohydr Res 211:91–102
Evans LR, Linker A (1973) Production and characterization of the slime polysaccharide of Pseudomonas aeruginosa. J Bacteriol 116:915–924
Gacesa P (1988) Alginates. Carbohydr Polym 8:161–182
Gorin PAJ, Spencer JFT (1966) Exocellular alginic acid from Azotobacter vinelandii. Can J Chem 44:993–998
Haug A, Larsen B, Smidsrød O (1966) A study of the constitution of alginic acid by partial acid hydrolysis. Acta Chem Scand 20:183–190
Haug A, Larsen B, Smidsrød O (1967) Studies on the sequence of uronic acid residues in alginic acid. Acta Chem Scand 21:691–704
Horikoshi K, Akiba T (1982) Alkalophilic microorganisms. Japan Scientific Societies Press, Tokyo, p 137
Iwamoto Y, Araki R, Iriyama K, Oda T, Fukuda H, Hayashida S, Muramatsu T (2001) Purification and characterization of bifunctional alginate lyase from Alteromonus sp. strain No. 272 and its action on saturated oligomeric substrates. Biosci Biotechnol Biochem 65:133–142
Kawamoto H, Horibe A, Miki Y, Kimura T, Tanaka K, Nakagawa T, Kawamukai M, Matsuda H (2006) Cloning and sequencing analysis of alginate lyase genes from the marine bacterium Vibrio sp. O2. Mar Biotechnol 8:481–490
Kitamikado M, Tseng CH, Yamaguchi K, Nakamura T (1992) Two types of alginate lyases. Appl Environ Microbiol 58:2474–2478
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lange B, Wingender J, Winkler UK (1989) Isolation and characterization of an alginate lyase from Klebsiella aerogenes. Arch Microbiol 152:302–308
Matsubara Y, Kawada R, Iwasaki K, Oda T, Muramatsu T (1998) Extracellular poly (α-l-guluronate) lyase from Corynebacterium sp.: purification, characterization, and conformational properties. J Protein Chem 17:29–36
Min KH, Sasaki SF, Kashiwabara Y, Nishizawa K (1977) Substrate specificity of endo-polyguluronide lyase of Pseudomonas sp. J Biochem 81:547–553
Muramatsu T, Hashimoto H, Takahashi T (1984) Physicochemical characteristics and conformational features of alginate lyase isozymes form Turbo cornutus. Biosci Biotechnol Biochem 48:79–85
Osawa T, Matsubara Y, Muramatsu T, Kimura M, Kakuta Y (2005) Crystal structure of the alginate (poly-α-l-guluronate) lyase from Corynebacterium sp. at 1.2 Å resolution. J Mol Biol 345:1111–1118
Pereira L, Sousa A, Coelho H, Amado AM, Ribeiro-Claro PJA (2003) Use of FTIR, FT-Raman and 13C-NMR spectroscopy for identification of some seaweed phycocolloids. Biomol Eng 20:223–228
Saito H, Miura K (1963) Preparation of transforming deoxyribonucleic acid by phenol treatment. Biochim Biophys Acta 72:619–629
Sartori C, Finch DS, Ralph B (1997) Determination of the cation content of alginate thin films by FT i.r. spectroscopy. Polymer 38:43–51
Shirai T, Kobayashi T, Ito S, Horikoshi K (2008) Chapter IV-Alkaline adaptation of proteins. In: Siddiqui KS, Thomas T (eds) Protein adaptation in extremophiles. Nova Science Publishers Inc, Hauppauge, NY, pp 105–141
Suda K, Tanji Y, Hori K, Unno H (1999) Evidence for a novel Chlorella virus-encoded alginate lyase. FEMS Microbiol Lett 180:45–53
Sugimura I, Sawabe T, Ezura Y (2000) Cloning and sequence analysis of Vibrio halioticoli genes encoding three types of polyguluronate lyase. Mar Biotechnol 2:65–73
Suzuki H, Suzuki K, Inoue A, Ojima T (2006) A novel oligoalginate lyase from abalone, Haliotis discus hannai, that releases disaccharide from alginate polymer in an exolytic manner. Carbohydr Res 341:1809–1819
Svanem BIG, Strand WI, Ertesvåg H, Skjåk-Bræk G, Hartmann M, Barbeyron T, Valla S (2001) The catalytic activities of the bifunctional Azotobacter vinelandii mannuronan C-5-epimerase and alginate lyase AlgE7 probably originate from the same active site in the enzyme. J Biol Chem 276:31542–31550
Weissbach A, Hurwitz J (1959) The formation of 2-keto-3-deoxyheptonic acid in extracts of Escherichia coli B. J Biol Chem 234:705–709
Wong TY, Preston LA, Schiller NL (2000) Alginate lyase: review of major sources and enzyme characteristics, structure-function analysis, biological roles, and applications. Ann Rev Microbiol 54:289–340
Xiao L, Han F, Yang Z, Lu XZ, Yu WG (2006) A novel alginate lyase with high activity on acetylated alginate of Pseudomonas aeruginosa FRD1 from Pseudomonas sp. QD03. World J Microbiol Biotechnol 22:81–88
Yamasaki M, Moriwaki S, Miyake O, Hashimoto W, Murata K, Mikami B (2004) Structure and function of a hypothetical Pseudomonas aeruginosa protein PA1167 classified into family PL-7. J Biol Chem 279:31863–31872
Yamasaki M, Ogura K, Hashimoto W, Mikami B, Murata K (2005) A structural basis for depolymerization of alginate by polysaccharide lyase family-7. J Mol Biol 352:11–21
Acknowledgments
We are grateful to the captain and crew of the R/V Natushima and the ROV Hyper Dolphin operation team, for their technical support in sampling. We also thank the chief scientist, Prof. K. Kubokawa of the Ocean Research Institute, University of Tokyo, as well as all scientists aboard the NT05-12 cruise. We thank Dr. S. Aihara and Dr. H. Minegishi of Bio-Nano Electronics Research Center, Toyo University, for the IR spectra.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by L. Huang.
Rights and permissions
About this article
Cite this article
Kobayashi, T., Uchimura, K., Miyazaki, M. et al. A new high-alkaline alginate lyase from a deep-sea bacterium Agarivorans sp.. Extremophiles 13, 121–129 (2009). https://doi.org/10.1007/s00792-008-0201-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-008-0201-7