
Abstract
All members of the genera Haloarcula and Halomicrobium whose names have been validly published were surveyed for 16S rRNA gene polymorphism, and the transcription of the genes from two species was investigated during growth at different NaCl concentrations. The species of Haloarcula and Halomicrobium harbour at least two different 16S rRNA gene copies, and 18 new sequences of 16S rRNA genes were obtained. The type I and type II 16S rRNA genes of Haloarcula are divergent at 4.8–5.6% of their nucleotide positions. The type III and type IV 16S rRNA genes from Halomicrobium mukohataei JCM 9738T are 9.0% divergent, which represents the highest intraspecific divergent 16S rRNA genes so far seen. Phylogenetic analysis based on 16S rRNA genes indicated that all type I 16S rRNA genes were clustered, and the same was true for the type II 16S rRNA genes of Haloarcula species. The two clusters, respectively generated from type I and type II 16S rRNA genes, were sharply separated and their divergences (4.8–5.6%) are in the range of various divergence usually found between genera in the order Halobacteriales (about 5–10%). Results from reverse transcription-PCR showed that the type I and type II copies of Har. amylolytica BD-3T and type III and type IV copies of Hmc. mukohataei JCM 9738T were all transcribed to 16S rRNA molecules under different salt concentrations (15–28% NaCl).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The gene coding for the small subunit ribosomal RNA (SSU) has been an important molecular chronometer for identification and classification of prokaryotes (Woese et al. 1990) and is now being extensively used to evaluate prokaryotic diversity in natural environments (Case et al. 2007). However, some prokaryotic species harbour several divergent 16S rRNA genes in their genomes, which cause concern about the reliability of 16S rRNA gene analysis in the classification and identification of prokaryotes, as well as in the evaluation of prokaryotic diversity (Clayton et al. 1995; Wang et al. 1997; Yap et al. 1999; Marchandin et al. 2003). Within the domain Archaea, several methanogenic species such as Methanocaldococcus jannaschii, Methanospirillum hungatei and Methanothermobacter thermautotrophicus exhibit 16S rRNA gene polymorphisms displaying 0.1% divergence, while some halophilic archaea harbour more divergent 16S rRNA gene copies with 0.1–6.8% divergence. These include species of Haloarcula, Halosimplex carlsbadense (Vreeland et al. 2002), Natrinema sp. strain XA3-1 (Boucher et al. 2004), Haloquadratum walsbyi (Burns et al. 2007) and Haladaptatus paucihalophilus (Savage et al. 2007). Boucher et al. (2004) proposed that 16S rRNA gene polymorphisms are an evolutionarily stable characteristic of halophilic archaea.
The occurrence of different copies of 16S rRNA gene has been known for several members of Haloarcula, and transcription of each copy of the 16S rRNA genes is probably related to environmental fluctuations (Dennis 1999). In Haloarcula marismortui, the two types of 16S rRNA genes are present in approximately equal amounts in ribosomes, and both rRNA operons of Har. marismortui have been shown to be expressed during cultivation under standard laboratory growth conditions (Mylvaganam and Dennis 1992; Amann et al. 2000). Salinity fluctuations in hypersaline environments may have a significant influence on the physiology of halophiles (Dennis 1999; Dennis and Shimmin 1997), and the presence of two distinct ribosome populations may therefore allow Har. marismortui to maintain essential protein synthesis during the periods of environmental stress. In fact, three rRNA operons were found in the genome of Har. marismortui (Baliga et al. 2004), and all the three copies of 16S rRNA genes are transcribed in the wild-type Har. marismortui. However, a single rRNA operon mutant (rrnA, ΔrrnB, ΔrrnC) grows on rich media similar to the wild-type (Tu et al. 2005). Other members of Haloarcula such as Har. quadrata and Har. amylolytica have two copies of 16S rRNA genes which differ at 5.0% of their nucleotide positions, while Har. hispanica and Har. vallismortis harbour two types of 16S rRNA gene with 2.4 and 2.9% divergence, respectively. It was reported that Har. japonica possessed four copies of the 16S rRNA gene (Gemmell et al. 1998), whereas multicopy genes have not been reported for Har. argentinensis and Halomicrobium mukohataei (Ihara et al. 1997; Oren et al. 2002).
To better understand the 16S rRNA gene polymorphism in Haloarcula and the related genus Halomicrobium, we designed specific-primer-based PCR towards identifying specific gene copies in each species, and investigated the transcription of different types of 16S rRNA genes at different salinities in representative species of Haloarcula and Halomicrobium.
Materials and methods
Strains and growth conditions
Har. argentinensis JCM 9737T, Har. quadrata JCM 11048T, Har. japonica JCM 7785T and Hmc. mukohataei JCM 9738T were purchased from JCM (Japan Collection of Microorganisms, Wako, Japan). Har. hispanica CGMCC 1.2049T, Har. marismortui CGMCC 1.1784T and Har. vallismortis CGMCC 1.2048T were from CGMCC (China General Microbiological Culture Collection, Beijing, China). Har. amylolytica BD-3T was recently described (Yang et al. 2007).
Unless stated otherwise, cultures were grown aerobically with vigorous shaking at 37°C in a Complete Medium (CM medium) (Sehgal and Gibbons 1960), while for reverse transcription (RT)-PCR experiments, Har. amylolytica BD-3T and Hmc. mukohataei JCM 9738T were cultivated in CM media with different NaCl concentrations (15, 20, 25 and 28%, w/v).
Design of specific primers
All three 16S rRNA gene sequences of Har. marismortui ATCC 43049T were retrieved from its genome (NC_006396; NC_006397) and were used to design specific primers targeting the different 16S rRNA gene copies of Haloarcula, while the two 16S rRNA gene sequences of Hmc. mukohataei JCM 9738T (cloned and sequenced in this study) were used to design specific primers for Halomicrobium. These sequences were aligned using the ClustalW program in MEGA3.1 software (Kumar et al. 2004) and the most divergent regions from each were used for primers design (Table 1).
Identification of 16S rRNA gene polymorphism in Haloarcula and Halomicrobium
Genomic DNAs from all halophilic archaeal strains were prepared as described by Ng et al. (1995). Different copies of 16S rRNA genes in each species were surveyed by PCR amplification of individual copies of the 16S rRNA gene with the above-designed specific primers. PCR was performed in a thermal cycler (MJ Research PTC-200, USA) for 30 cycles (5 min denaturing step at 95°C in the first cycle; 0.5 min denaturing at 95°C, 0.5 min annealing at 55°C and 0.5 min elongation at 72°C, with a final extension step at 72°C for 5 min). The PCR products were examined on a 1.5% agarose gel.
Amplification of 16S rRNA genes with universal primers and phylogenetic analysis
The universal oligonucleotide primers designed for amplification of the haloarchaeal 16S rRNA genes were the forward primer 0018F (5′-ATTCCGGTTGATCCTGCC-3′) and the reverse primer 1518R (5′-AGGAGGTGATCCAGCCGC-3′), designed based on the upstream and downstream sequences of the 16S rRNA genes of all species of Halobacteriaceae. The amplified products were recovered from the agarose gel by using a gel extraction kit (Tiangen, China), and were cloned into the pGEM-T vector (Promega, USA) and transformed into Escherichia coli TOP 10. Positive clones harbouring cloned 16S rRNA gene fragments were selected by PCR using specific primers (type I–type IV, Table 1) and were sequenced by SinoGenoMax Company Limited (Beijing, China). All sequences were checked for possible chimeric artefacts by the Chimera Detection program in the Ribosomal Database Project II (Cole et al. 2003). The nucleotide sequences determined in this study have been deposited in the GenBank database under accession numbers EF645680–EF645694. Multiple sequence alignments were performed using the ClustalW program integrated in MEGA3.1 software. Phylogenetic trees were reconstructed using the Neighbour-Joining and Maximum-Parsimony methods (Saitou and Nei 1987) with the Kimura 2-parameter calculation model (Kimura 1980) in MEGA3.1 software. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown next to the branches (Felsenstein 1985).
RT-PCR and identification of different 16S rRNAs
Mid-exponential phase cultures of Har. amylolytica BD-3Tand Hmc. mukohataei JCM 9738T cultivated in CM medium with different NaCl concerntrations were harvested and total RNAs were extracted as previously described (Nieuwlandt et al. 1995).
For reverse transcription (RT), the reverse primer 1518R was used to prime the synthesis of cDNA. One microgram portion of total RNAs was used as template, and RT was carried out according to the instructions of the supplier (MMLV-RT, Epicentre Biotechnologies). The mock cDNA reaction where RT was not carried out, excluding the possibility of PCR amplification from residual genomic DNA, was used as the control. One microliter of the RT mixture was used directly as the template in subsequent PCRs. A 20-µl PCR mixture contained 10 pmol of specific primers, 200 mM of each dATP, dGTP, dCTP and dTTP, 1 U of Taq DNA polymerase (Promega), and 1× buffer provided with the Taq polymerase.
Results
Specificity of the newly designed primers
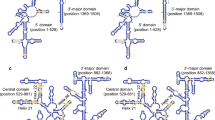
Four sets of primers that target the variable region of the 16S rRNA gene were designed. The sequences, target positions at the 16S rRNA gene, and the expected sizes of PCR products are listed in Table 1. The specificity of each primer pair was experimentally tested by using clones with a single 16S rRNA gene from either Har. amylolytica BD-3T or Hmc. mukohataei JCM 9738T (Fig. 1). When 16S rRNA gene-specific amplification was performed with the newly designed primers, PCR products of the expected size were obtained. Each pair of primers was shown to amplify a fragment of the expected size only from its corresponding 16S rRNA gene and did not cross-react with any of the non-targeted 16S rRNA genes.

Specificity of primers for detection of two types of 16S rRNA genes in Har. amylolytica BD-3T (left) and Hmc. mukohataei JCM 9738T (right). Lanes M-DNA marker, lanes 1 and 4: Har. amylolytica BD-3T rrnA, lanes 2 and 5: Har. amylolytica BD-3T rrnB, lanes 3 and 6: Har. amylolytica BD-3T rrnC, lanes 1′ and 2′: Hmc. mukohataei JCM 9738T rrnA, lanes 2′ and 4′: Hmc. mukohataei JCM 9738T rrnB

Neighbour-Joining phylogenetic tree based on 16S rRNA gene sequences of all members of Haloarcula, Halomicrobium. Bootstrap values (%) are based on 1,000 replicates and are shown for branches with more than 70% bootstrap support. Bar represents 0.01 expected changes per site. The boldfaced species were newly sequenced in this study
Polymorphism of 16S rRNA genes and their influence on phylogenetic analysis of the species of Haloarcula and Halomicrobium
By using the designed 16S rRNA gene-specific primers and extracted genomic DNAs as templates, the polymorphism of 16S rRNA genes in species of the genera Haloarcula and Halomicrobium were studied. At least two different 16S rRNA genes were detected in seven Haloarcula species and also in the sole Halomicrobium species, Hmc. mukohataei. Each of the different types of 16S rRNA gene copies was sequenced, and 18 new sequences of 16S rRNA genes were obtained (GenBank Acc. Nos. EF645680–EF645694, DQ826512–DQ826513, DQ854818). The overall size of the amplified fragments of 16S rRNA genes of Haloarcula species was 1472 bp, while the two 16S rRNA gene fragments of Hmc. mukohataei JCM 9738T were 1473 bp (rrnA) and 1472 bp (rrnB), respectively. Sequence alignments and analyses of the 16S rRNA genes of Haloarcula species showed that these genes formed two stable clusters (Fig. 1). The genes that clustered with rrnA and rrnB or rrnC of Har. marismortui ATCC 43049T were, respectively, designated as type I and type II 16S rRNA genes. Alignment of the type I and type II 16S rRNA gene sequences of all Haloarcula species revealed 4.8–5.6% nucleotide substitutions. The rrnA and rrnB of Hmc. mukohataei JCM 9738T were, respectively, designated as type III and type IV 16S rRNA genes, and they showed 9.0% divergence, representing the highest intraspecific divergent 16S rRNA genes so far known.
Detection of various types of 16S rRNA molecules by RT-PCR
To examine whether all the 16S rRNA genes from Haloarcula and Halomicrobium species were transcribed and whether their transcription was regulated by the NaCl concentration, total RNAs were prepared from cultures grown in CM medium containing different concentrations of NaCl. The cDNAs were first synthesized from the total RNAs by RT, using the universal primer 1518R that was complementary to the 3′ end of both type I and type II, or type III and type IV 16S rRNAs. Second, the specific PCR primers (Table 1) were used, so that either type I, type II, type III, or type IV c DNAs of 16S rRNA fragments were amplified. The results are shown in Figs. 3 and 4. When the cDNAs were used as the template in PCR, all pairs of primers (I–IV, Table 1) amplified fragments of the correct size. None of the primers amplified products from the mock cDNA reaction where RT was omitted, which excluded the possibility of PCR amplification from genomic DNA contaminants. The RT-PCR experiment clearly demonstrated that both type I and type II 16S rRNA genes were transcribed in Har. amylolytica. Although these experiments did not quantify the transcription of each type of 16S rRNA gene, the amounts of the type I and type II 16S rRNAs from cells cultivated at different NaCl concentrations were comparable as judged by the intensities of the bands of PCR-amplified fragments, indicating that the type I or type II genes were transcribed at all tested NaCl concentrations. The type III and type IV 16S rRNA genes of Hmc. mukohataei were also transcribed at all NaCl concentrations (Fig. 4).

Detection of type I and type II 16S rRNAs in Har. amylolytica BD-3T cultivated in CM medium at different concentrations of NaCl by RT-PCR. Lanes M-DNA marker, lanes 1, 2, 9 & 10: cultivated in CM medium with 15% NaCl, lanes 3, 4, 11 and 12: cultivated in CM medium with 20% NaCl, lanes 5, 6, 13 and 14: cultivated in CM medium with 25% NaCl, lanes 7, 8, 15 and 16: cultivated in CM medium with 28% of NaCl

Detection of type III and type IV 16S rRNAs in Hmc. mukohataei JCM 9738T cultivated in CM medium at different concentrations of NaCl by RT-PCR. Lanes M-DNA marker, lanes 1, 2, 9 and 10: cultivated in CM medium with 15% NaCl, lanes 3, 4, 11 and 12: cultivated in CM medium with 20% NaCl, lanes 5, 6, 13 and 14: cultivated in CM medium with 25% NaCl, lanes 7, 8, 15 and 16: cultivated in CM medium with 28% of NaCl
Discussion
In the domains Eukarya and Bacteria, the presence of two distinct types of SSU genes in a single genome has been reported to be an isolated event that affects only a few species such as two thermophilic actinomycetes, Thermomonospora chromogena and Thermobispora biopsora (Wang et al. 1997; Yap et al. 1999), Vibrio species (Harth et al. 2007), the apicomplexan Plasmodium berghei (Gunderson et al. 1987) and the metazoan Dugesia mediterranea (Carranza et al. 1996). As to halophilic archaea, 16S rRNA gene polymorphism may be a widespread feature. Here, we present data on the extent of 16S rRNA gene polymorphism for all species of Haloarcula and Halomicrobium whose names have been validly published. We have confirmed that four types (type I–type IV) of 16S rRNA genes occur in Haloarcula and Halomicrobium. Type I and type II were detected in Haloarcula and they were actively transcribed at NaCl concentrations of 15–28%. Type III and type IV were found in Halomicrobium and they were both transcribed at different NaCl concentrations. Differential transcription of the two genes of Har. marismortui has been shown to be temperature-dependent (López-López et al. 2007).
The type I and type II 16S rRNA genes of Haloarcula fall into two monophyletic clusters (cluster I and cluster II in Fig. 2), suggesting two distinct evolutionary lineages in each species. The clustering of these two related monophyletic groups indicates that all species of Haloarcula might have speciated from a common ancestral organism in which the divergent copies of 16S rRNA genes had already existed in the genome. Hmc. mukohataei JCM 9738T also harbours two different 16S rRNA genes (type III and type IV). Of the two 16S rRNA genes of Hmc. mukohataei, type III (16S rrnA) is more related to Haloarcula groups based on the sequence similarity, while the IV (16S rrnB) is more distant in the tree. The type III and type IV genes of Hmc. mukohataei showed 9% divergence. This is the highest value so far known.
The degree of intraspecies divergences observed for the 16S rRNA genes of Haloarcula (more than 5.0%) and Halomicrobium (9.0%) is far greater than the average interspecies divergence (~2.0%) and is more in the range of the divergence usually found between different genera in the family Halobacteriaceae (about 5–10%) (Boucher et al. 2004). This large divergence may cause concerns on the identification of haloarchaeal species based on 16S rRNA gene sequence. Historically, one example of taxonomic confusion possibly caused by intragenomic SSU gene heterogeneity is the status of the genera Haloterrigena and Natrinema (Boucher et al. 2004). This current study clearly demonstrated that two types of 16S rRNA genes occur in Haloarcula species and that all Haloarcula species fall into two monophyletic clusters corresponding to the type I and type II 16S rRNA genes. In-depth phylogenetic analysis would avoid potential confusion on identification of potential Haloarcula species only when a single 16S rRNA gene sequence is available. Ideally, all copies of 16S rRNA genes need to be sequenced, and the specific primers designed in this study could help obtaining different types of 16S rRNA genes from a single genome. When considering the effects of this heterogeneity on the accuracy of species diversity estimation by 16S rRNA-based methods, it is conceivable that analysis of environmentally originated 16S rRNA gene pools may lead to an overestimation of the halophilic archaeal diversity. Therefore, the 16S rRNA genes should be used with caution as an indicator in studies of environmental haloarchaeal biodiversity (Crosby and Criddle 2003).
References
Amann G, Stetter KO, Llobet-Brossa E, Amann R, Antón J (2000) Direct proof for the presence and expression of two 5% different 16S rRNA genes in individual cells of Haloarcula marismortui. Extremophiles 4:373–376
Baliga NS, Bonneau R, Facciotti MT, Pan M, Glusman G, Deutsch EW, Shannon P, Chiu Y, Weng RS, Gan RR, Hung P, Date SV, Marcotte E, Hood L, Ng WV (2004) Genome sequence of Haloarcula marismortui: a halophilic archaeon from the Dead Sea. Genome Res 14:2221–2234
Boucher Y, Douady CJ, Sharma AK, Kamekura M, Doolittle WF (2004) Intragenomic heterogeneity and intergenomic recombination among haloarchaeal rRNA genes. J Bacteriol 186:3980–3990
Burns DG, Janssen PH, Itoh T, Kamekura M, Li Z, Jensen G, Rodríguez-Valera F, Bolhuis H, Dyall-Smith ML (2007) Haloquadratum walsbyi gen. nov., sp. nov., the square haloarchaeon of Walsby, isolated from saltern crystallizers in Australia and Spain. Int J Syst Evol Microbiol 57:387–392
Carranza S, Giribet G, Riberat C, Baguna J, Riutort M (1996) Evidence that two types of 18S rDNA coexist in the genome of Dugesia (Schmidtea) mediterranea (Platyhelminthes, Turbellaria, Tricladida). Mol Biol Evol 13:824–832
Case RJ, Boucher Y, Dahllöf I, Holmström C, Doolittle WF, Kjelleberg S (2007) Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl Environ Microbiol 73:278–288
Clayton RA, Sutton G, Hinkle PS Jr, Bult C, Fields C (1995) Intraspecific variation in small-subunit rRNA sequences in GenBank: why single sequences may not adequately represent prokaryotic taxa. Int J Syst Bacteriol 45:595–599
Cole JR, Chai B, Marsh TL, Farris RJ, Wang Q, Kulam SA, Chandra S, McGarrell DM, Schmidt TM, Garrity GM, Tiedje JM (2003) The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res 31:442–443
Crosby LD, Criddle CS (2003) Understanding bias in microbial community analysis techniques due to rrn operon copy number heterogeneity. Biotechniques 34:790–794
Dennis PP (1999) Expression of ribosomal RNA operons in halophilic archaea. In: Oren A (ed) Microbiology and biogeochemistry of hypersaline environments. CRC Press, Boca Raton, pp 319–329
Dennis PP, Shimmin LC (1997) Evolutionary divergence and salinity-mediated selection in halophilic archaea. Microbiol Mol Biol Rev 61:90–104
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Gemmell RT, McGenity TJ, Grant WD (1998) Use of molecular techniques to investigate possible long-term dormancy of halobacteria in ancient halite deposits. Ancient Biomol 2:125–133
Gunderson JH, Sogin ML, Wollett G, Hollingdale M, de la Cruz VF, Waters AP, McCutchan TF (1987) Structurally distinct, stage-specific ribosomes occur in Plasmodium. Science 238:933–937
Harth E, Romero J, Torres R, Espejo RT (2007) Intragenomic heterogeneity and intergenomic recombination among Vibrio parahaemolyticus 16S rRNA genes. Microbiology 153:2640–2647
Ihara K, Watanabe S, Tamura T (1997) Haloarcula argentinensis sp. nov. and Haloarcula mukohataei sp. nov., two new extremely halophilic archaea collected in Argentina. Int J Syst Bacteriol 47:73–77
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kumar S, Tamura K, Nei M (2004) MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
López-López A, Benlloch S, Bonfá M, Rodríguez-Valera F, Mira A (2007) Intragenomic 16S rDNA divergence in Haloarcula marismortui is an adaptation to different temperatures. J Mol Evol 65:687–696
Marchandin H, Teyssier C, Simeon De Buochberg M, Jean-Pierre H, Carriere C, Jumas-Bilak E (2003) Intra-chromosomal heterogeneity between the four 16S rRNA gene copies in the genus Veillonella: implications for phylogeny and taxonomy. Microbiology 149:1493–1501
Mylvaganam S, Dennis PP (1992) Sequence heterogeneity between the two genes encoding 16S rRNA from the halophilic archaebacterium Haloarcula marismortui. Genetics 130:399–410
Ng WL, Yang CF, Halladay JT, Arora P, DasSarma S (1995) Protocol 25: isolation of genomic and plasmid DNAs from Halobacterium halobium. In: DasSarma S, Fleischmann EM (eds) Archaea: a laboratory manual: halophiles. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 179–180
Nieuwlandt DT, Palmer JR, Armbruster DT, Kuo YP, Oda W, Daniels CJ (1995) Protocol 23: a rapid procedure for the isolation of RNA from Haloferax volcanii. In: DasSarma S, Fleischmann EM (eds) Archaea: a laboratory manual: halophiles. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 161–162
Oren A, Elevi R, Watanabe S, Ihara K, Corcelli A (2002) Halomicrobium mukohataei gen. nov., comb. nov., and emended description of Halomicrobium mukohataei. Int J Syst Evol Microbiol 52:1831–1835
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Savage KNL, Krumholz LR, Oren A, Elshahed MS (2007) Haladaptatus paucihalophilus gen. nov., sp. nov., a halophilic archaeon isolated from a low-salt, sulfide-rich spring. Int J Syst Evol Microbiol 57:19–24
Sehgal SN, Gibbons NE (1960) Effect of some metal ions on the growth of halobacterium cutirubrum. Can J Microbiol 6:165–169
Tu D, Blaha G, Moore PB, Steitz TA (2005) Gene replacement in Haloarcula marismortui: construction of a strain with two of its three chromosomal rRNA operons deleted. Extremophiles 9:427–435
Vreeland RH, Straight S, Krammes J, Dougherty K, Rosenzweig WD, Kamekura M (2002) Halosimplex carlsbadense gen. nov., sp. nov., a unique halophilic archaeon, with three 16S rRNA genes, that grows only in defined medium with glycerol and acetate or pyruvate. Extremophiles 6:445–452
Wang Y, Zhang ZS, Ramanan N (1997) The actinomycete Thermobispora bispora contains two distinct types of transcriptionally active 16S rRNA genes. J Bacteriol 179:3270–3276
Woese CR, Kandler O, Wheelis ML (1990) Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 87:4576–4579
Yang Y, Cui HL, Zhou PJ, Liu SJ (2007) Haloarcula amylolytica sp. nov., an extremely halophilic archaeon isolated from Aibi salt lake in Xin-Jiang, China. Int J Syst Evol Microbiol 57:103–106
Yap WH, Zhang Z, Wang Y (1999) Distinct types of rRNA operons exist in the genome of the actinomycete Thermomonospora chromogena and evidence for horizontal transfer of an entire rRNA operon. J Bacteriol 181:5201–5209
Acknowledgments
This study was supported by the National Basic Research Program from the Ministry of Science and Technology (MOST) of China (Grant No. 2004CB719601) and a start-up grant from Jiangsu University (Grant No. 08JDG016).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Driessen.
Rights and permissions
About this article
Cite this article
Cui, HL., Zhou, PJ., Oren, A. et al. Intraspecific polymorphism of 16S rRNA genes in two halophilic archaeal genera, Haloarcula and Halomicrobium . Extremophiles 13, 31–37 (2009). https://doi.org/10.1007/s00792-008-0194-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-008-0194-2