
Abstract
In contrast to conventional wastewater treatment plants and saline environments, little is known regarding the microbial diversity of hypersaline wastewater. In this study, the microbial communities of a hypersaline tannery effluent, and those of three treatment systems operating with the tannery effluent, were investigated using 16S rDNA phylogenetic markers. The comparative analysis of 377 bacterial sequences revealed the high diversity of this type of hypersaline environment, clustering within 193 phylotypes (≥ 97% similarity) and covering 14 of the 52 divisions of the bacterial domain, i.e. Proteobacteria, Bacteroidetes, Firmicutes, Actinobacteria, Chlorobi, Planctomycetes, Spirochaetes, Synergistes, Chloroflexi, Thermotogae, Verrucomicrobia, OP3, OP11 and TM7. Most of the phylotypes were related to halophilic and pollutant-degrading bacteria. Using statistical analysis, the diversity of this type of environment was compared to that of other environmental samples selected on the basis of their salinity, oxygen content and organic load.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The interest in halophilic microorganisms is largely motivated not only by the industrial application of these microbes, notably in the production of enzymes (Jones 2004) and exopolysaccharides (Quesada et al. 2004), but also for their use in depollution systems (Peyton et al. 2004). Regarding this last application, pollution removal in hypersaline wastewater is likely to represent up to 5% of the global wastewater treatment requirement. Although the number of studies dealing with the biological treatment of hypersaline wastewater is increasing rapidly, little is known regarding the diversity of these halophilic communities.
It has already been shown that high salinity permits the growth of a complex and diverse halophilic microflora in several types of saline water, such as seawater (Venter et al. 2004), salterns (Anton et al. 1999, 2000; Benlloch et al. 1995; Martinez-Murcia et al. 1995; Rodríguez-Valera et al. 1999) and soda lakes (Duckworth et al. 1996; Hollibaugh et al. 2001; Litchfield and Gillevet 2002; Sorokin and Kuenen 2005; Ward et al. 2000). Furthermore, sludge diversity in conventional biological wastewater treatment processes, both anaerobic and aerobic, has also been characterised and appears to be very broad, though different from that in fresh water (Bond et al. 1995; Godon et al. 1997a, b; Snaidr et al. 1997). However, only scanty knowledge is available regarding the microbial diversity of biological wastewater treatment systems operating with a high salt content. Yet, it has already been shown that the diversity of a salt-tolerant ecosystem treating hypersaline industrial wastewater could be similar to the diversity of a non-salt-tolerant one (Lefebvre et al. 2004).
In this study, we propose to describe the bacterial diversity in terms of species richness associated with a highly saline wastewater, i.e. tannery effluent, and different treatment plants used to clean this pollution. Leather tanning is almost wholly a wet process from which a large volume of highly saline liquid waste is continuously generated. The tanning process and the effluents generated have already been reported in the literature (Sreeram and Ramasami 2003; Stoop 2003; Wiegant et al. 1999). The aim of this study was firstly to describe the microbial communities involved in the aerobic and anaerobic treatment of hypersaline wastewater; and secondly, to compare the diversity potential of such wastewater to that of biological sludge involved in the aerobic and anaerobic treatment of non-saline wastewater, taking into account the saline diversity.
Materials and methods
Sampling
Four different samples were collected for this experiment: first, tannery soak liquor (TSL) was collected from soak pits in a tannery located in a tannery cluster situated in the area of Chennai, India. Second, activated sludge was collected from a common effluent treatment plant (CETP) in the same area, treating the composite effluents of 128 tanneries and minor amounts of domestic wastewater. The third and fourth samples collected were biological sludge from two laboratory-scale bioreactors run with TSL: one aerobic sequencing batch reactor (SBR) and one upflow anaerobic sludge blanket reactor (UASB). The salt (NaCl) content of these samples were 72.3, 6.9, 52.5 and 45.6 g l−1 for the TSL, CETP, SBR and UASB, respectively.
Total extraction, amplification, cloning and sequencing of genomic 16S rDNA
Total genomic DNA was extracted and purified from 10 ml of sample using a QiaAmp DNA stool mini kit, in accordance with the manufacturer’s instructions (Qiagen, Hilden, Germany).
After extraction, bacterial 16S rDNA genes were amplified by PCR using the bacterial forward primer W18 (5′-GAGTTTGATCMTGGCTCAG-3′) and the universal reverse primer W31 (5′-TTACCGCGGCTGCTGGCAC-3′), previously described by Snell-Castro et al. (2005), in positions 9–27 and 482–500 in Escherichia coli, respectively (Brosius et al. 1981). 16S rDNA genes from the archaeal domain were targeted using the archaeal forward primer W17 (5′-ATTCYGGTTGATCCYGSCRG-3′) and the universal reverse primer W02 (5′-GNTACCTTGTTACGACTT-3′), previously described by Godon et al. (1997b), E. coli positions 3–22 and 1509–1492, respectively. Eukaryotic forward primer W99 (5′-CGGTAATTCCAGCTCC-3′) (Edgcomb et al. 2002), E. coli positions 528–544, and W02 were used for the partial amplification of 18S rRNA eukaryotic genes. PCR reactions were as follows: each PCR reaction tube contained 2 μl of each primer at a concentration of 100 ng μl−1, along with 5 μl of 1X Taq reaction buffer (Perkin Elmer, Forster City, CA, USA), 4 μl of dNTP 2.5 mM, 1 μl of Taq DNA polymerase (Perkin Elmer) and water, for a final volume of 49 μl. This PCR mix was then added to 1 μl of DNA diluted five times in water. PCR conditions were as follows: an initial denaturation step at 94°C for 2 min, followed by 25 cycles of a three-stage program with 1 min at 94°C, 1 min at 55°C and 1 min at 72°C, the final elongation step running for 10 min at 72°C. PCR products were purified with Qiagen microcolumns, in accordance with the manufacturer’s instructions (Qiagen). Purified PCR products were cloned and transformed into TOP10 E. coli competent cells using the PCR 4-TOPO vector kit, according to supplier instructions (Invitrogen, Groningen, the Netherlands). Recombinant cells were selected using kanamycine resistance and ccd gene killer inactivation. Afterwards, 96 individual clones were inoculated onto a plate containing YT medium/5% glycerol/ampicilline and sent for sequencing after 24 h incubation at 37°C.
Sequence analysis
16S rDNA sequences (of about 500 bp) were identified by comparison with sequences available in databases using the BLAST program (Altschul et al. 1997). To confirm phylum position, all the sequences were fitted into an alignment of about 12,000 full and partial 16S and 18S rRNA sequences using the automated tools of the ARB software package (Ludwig et al. 2004). Partial sequence data were incorporated into trees by neighbour-joining (Saitou and Nei 1987). Potential DNA chimeric structure was searched for by performing fractional treeing on the 5′ and 3′ ends of the sequenced DNA fragments and by analysis of the suspected sequences by the CHECK CHIMERA program (RDP-II) (Cole et al. 2003).
Nomenclature
The first letter of each clone name (e.g. MN100) corresponds to the set of primers used for its amplification; the second letter corresponds to an independent PCR amplification event; and the numeral corresponds to the clone number of the respective library. Each phylotype was usually named according to the first clone identified displaying the representative sequence. An acronym was also chosen in order to facilitate the identification of our samples, i.e. TERSS, standing for tannery effluent-related saline sludge. Thus, our samples will usually be referred to as TERSS-TSL for the tannery effluent-related saline sludge TSL sample, TERSS-SBR for the tannery effluent-related saline sludge–sequencing batch reactor sample, TERSS-UASB for the tannery effluent-related saline sludge upflow anaerobic sludge blanket sample and TERSS-CETP for the tannery effluent-related saline sludge CETP sample. The nucleotide sequence data reported in this work will appear in the GenBank nucleotide database under accession numbers AM157453 to AM157645.
Statistical analysis and sequence population diversity
Coverage was determined using a non-parametric estimator of the proportion of phylotypes in a library of infinite size that would be represented in a smaller library. Good’s coverage estimator (C Good) was calculated using the following formula: C Good = 1 − (n 1/N), where n 1 is the number of phylotypes that appeared only once in the clone library and N is the total number of clones analysed (Good 1953). Rarefaction curves and the Chao’s abundance-based coverage estimator of species richness (S ACE) (Chao et al. 2000; Chazdon et al. 1998) were produced by using the EstimateS software package for species richness calculation (Colwell 2005). Finally, a dendrogram was built up and a principal component analysis (PCA) was performed using the XLSTAT software package. The dendrogram was built up using the aggregation criteria of Ward, based on Euclidean distances, and the PCA was performed using the correlation coefficient of Pearson.
Results
Origin of the tannery wastewater sample
In the leather industry, salt (sodium chloride, NaCl) is used to preserve the fresh skins from decomposition immediately after they are stripped in the slaughterhouse, and subsequently, the excess of salt has to be removed in the tannery before further processing. This is done by soaking in large quantities of water, which generates the first source of effluent. This TSL (sample called TERSS-TSL) is characterised by its high organic load, large amount of suspended solids (sand, lime, hair, flesh, dung, etc.) and high salinity. A similar TSL sample had already been treated at bench scale—aerobically in a sequencing batch reactor (sample called TERSS-SBR) (Lefebvre et al. 2005), and anaerobically in an upflow anaerobic sludge blanket (sample called TERSS-UASB) (Lefebvre et al. 2006). In addition, the microbial diversity of activated sludge from an industrial-scale CETP—shared by 128 tanneries, treating tannery composite wastewater (i.e. the effluent made up of all the streams generated by a tannery, mixed together)—was also characterised (sample called TERSS-CETP). The microbial diversity of these four samples taken from a tannery environment was investigated using 16S rDNA analysis.
Global analysis of the 16S rDNA clone library
Archaeal 16S and eukaryotic 18S rDNA amplification was attempted using archaeal and eukaryotic specific primers and various PCR conditions. However, the amplification failed, which indicates that Archaea and Eukaryota, if present, were not major components of the autochthonous microbial community. Eighteen phyla among the 52 described by Rappé and Giovannoni were identified, as can be seen in Table 1 (Rappé and Giovannoni 2003). With regard to bacterial lineage, the microbial ecosystem observed by 16S rDNA gene comparative sequence analysis appeared to be diverse. The 377 bacterial sequences analysed clustered within 193 phylotypes, on the basis of at least 97% sequence similarity. The phylum most represented was that of Gammaproteobacteria, which contained 28% of all the sequences analysed and 20% of the phylotypes (see Table 1). Other main phyla were Bacteroidetes (20% of the sequences and 21% of the phylotypes), Firmicutes (19% of the sequences and 17% of the phylotypes) and Alphaproteobacteria (10% of the sequences and 15% of the phylotypes). The other sequences were distributed within 14 phyla representing 23% of the sequences and 25% of the phylotypes.
Most of the sequences recovered from the saline samples were not closely related to sequences present in public databases. From the 193 bacterial phylotypes observed, only 54 (28%) presented more than 97% similarity with the previously identified sequences. Seventy-nine (41%) showed similarity values in the range of 97–90%; the 60 others (31%) had similarity values between 90 and 80%. The sequences presenting more than 97% similarity are reported in Table 2. Only 100 phylotypes (52%) have been identified at the species level and were widely spread throughout the domain of Bacteria. The most abundant phylotype identified through this study was related to Halomonas alimentaria (AF211860) (MT100, 4% of the bacterial sequences), isolated from a traditional Korean fermented seafood. The phylogenetic diversity of each sample, based on phylum and phylotype levels, varied widely from one sample to another (see Tables 1, 2), justifying a separate analysis of each microbial ecosystem.
Comparative analysis of the TERSS-16S rDNA clone libraries
It can be seen in Table 2 that the percentage of phylotypes presenting more than 97% similarity with the previously identified phylotypes was only 11, 31, 41 and 43% (15, 31, 43 and 44% of the sequences) regarding the upflow anaerobic sludge blanket (TERSS-UASB), the CETP (TERSS-CETP), the aerobic sequencing batch reactor (TERSS-SBR) and the TSL (TERSS-TSL) samples, respectively. 38, 46, 41 and 40% of the phylotypes (37, 50, 42 and 44% of the sequences) showed similarity values in the range of 97–90%; the remaining 52, 23, 18 and 17% of the phylotypes (48, 19, 15 and 12% of the sequences) had similarity values between 90 and 80%. Regarding TERSS-UASB, only 6% of the phylotypes (11% of the sequences) were identified as already cultured species on the basis of at least 97% similarity. This proportion of sequences related to already cultured species was higher in the other samples reaching 23, 28 and 40% of the phylotypes (23, 21 and 42% of the sequences) in TERSS-SBR, TERSS-CETP and TERSS-TSL, respectively. Another criterion which was considered was the halotolerant or halophilic characteristics of our sequences: on the basis of at least 90% similarity, 16, 25, 55 and 71% of the phylotypes (18, 35, 63 and 82% of the sequences) of TERSS-UASB, TERSS-CETP, TERSS-SBR and TERSS-TSL, respectively, were related to species showing halophilic properties or to environmental clones isolated from saline environments. The phyla most represented in TERSS-TSL, TERSS-UASB and TERSS-SBR were Gammaproteobacteria, Bacteroidetes and Firmicutes, whereas TERSS-CETP was dominated by Alphaproteobacteria.
Coverage and similarity of libraries based on phylotype diversity
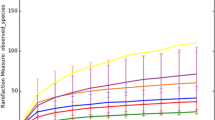
A deeper understanding of the microbial diversity was attained using various diversity indexes. The Good’s coverage values obtained indicated that the TERSS-CETP and the TERSS-UASB samples were more diverse than the TERSS-SBR and the TERSS-TSL samples, with C Good = 52, 54, 72 and 82%, respectively. The rarefaction curves obtained at a similar level of effort (Fig. 1) showed weakly curvilinear plots for the curves obtained for TERSS-CETP and TERSS-UASB samples, indicating that the phylotype richness in TERSS-CETP and TERSS-UASB environments was largely underestimated. The estimated number of phylotypes, determined using the Chao’s abundance-based coverage estimator of species richness (S ACE), was 50, 108, 144 and 231 phylotypes for TERSS-TSL, TERSS-SBR, TERSS-UASB and TERS-CETP, respectively. Finally, the similarities of the phylotypes in these libraries were represented in Fig. 2, in which it could be seen that the TERSS-SBR and TERSS-TSL 16 rDNA sequences showed around 30% similarity, whereas the other samples were very different. The TERSS-TSL and TERSS-CETP samples differed most, having no phylotype in common.

Rarefaction curves generated for 16S rDNA clone libraries from tannery effluent-related samples

Schematic representation of percentage similarities between four different samples related to tannery environment
Effect of salt, oxygen and organic load on phylum diversity
The similarity indexes previously calculated showed that, on the basis of phylotype diversity, all the samples were very different, apart from the aerobic sequencing batch reactor sample (TERSS-SBR) and the TSL sample (TERSS-TSL). Phylum level analysis seemed more suitable for a comparison of the four saline wastewater samples analysed here to other published environmental samples, using statistical tools. TERSS-SBR, TERSS-CETP and TERSS-TSL showed aerobic characteristics and can be compared to saline water according to their high salt content features, and to non-saline wastewater treatment sludge as regards their high organic load. In contrast, the TERSS-UASB sample displayed anaerobic characteristics and thus could be compared to non-saline anaerobic digestors. Therefore, the influence of environmental parameters on the diversity of our samples was studied using statistical analysis (dendrogram and PCA). Hence, our samples were clustered hierarchically into a dendrogram based on the phylum distribution among the samples, in order to present an analytic overview of the diversity. Additional reference data were used to build up the dendrogram, including descriptions of the microbial diversity of saline environments (seawater, salterns, soda lake), and also of conventional aerobic and anaerobic sludge from conventional biological treatment processes. Thus, a reference database was built up using 1,412 sequences from seawater (Venter et al. 2004), 197 from salterns (Benlloch et al. 2002; Mouné et al. 2003) 212 from soda lakes (Humayoun et al. 2003), 1,988 from anaerobic sludge (Godon et al. 2004, 1997b; Sekiguchi et al. 1998) and 256 from aerobic activated sludge (Bond et al. 1995; Snaidr et al. 1997).
All the samples clearly clustered in the dendrogram within three majors groups: the anaerobic sludge samples gathered in a first group, the aerobic sludge samples in a second group and the third group was composed of all the samples isolated from saline environments (data not shown). Our samples clustered in the third group, apart from TERSS-CETP that was clustered with the aerobic sludge. Among the three major parameters analysed (i.e. presence of oxygen, salt and organic load), salt thus appeared to distinguish clearly TERSS-SBR, TERSS-TSL and TERSS-UASB, whereas oxygen and organic load distinguished TERSS-CETP. A PCA analysis was then performed to further describe the relationship between our samples and the main types of reference ecosystems, as mentioned above. In this analysis, the environmental reference samples were pooled according to their environment and the dendogram results. It was therefore possible to explain 52% of the information with two axes, as shown in Fig. 3: axis 1 appears to be related to the amount of oxygen in the environment, separating samples into the aerobic on the left, the anaerobic on the right. In addition, all the samples appear to be aligned on axis 2 according to their salt concentration. According to Fig. 3, TERSS-SBR and TERSS-TSL clearly show saline properties. TERSS-UASB stands alone, in an intermediate position between the anaerobic sludge and saline samples, thus displaying characteristics from both. TERSS-CETP also stands alone, in an intermediate position between the conventional aerobic sludge and saline samples.

Principal component analysis carried out on four samples related to tannery wastewater (filled square) and on other reference samples characterising a specific type of environment (open square). The legend of the axis includes the percentage of diversity that is explained by each axis. The numerals in parentheses next to the sample names indicate the number of reference samples used to characterise the specific environments in the current figure
Discussion
Prokaryotic biodiversity of tannery effluent-related sludge communities
The tannery effluent-related sludge communities consisted mainly of organisms from the domain of Bacteria. In contrast, Archaea and Eukaryota could not actually be amplified. Though Archaea are seldom amplified in aerobic treatment processes, their absence seems surprising at the first sight in the UASB (TERSS-UASB), as methane production was observed in this digester, and had to be induced by methanogenic Archaea. However, at the salinity level of TERSS-UASB (i.e. 46 g NaCl l−1) halophilic Archaea amplification is known to be difficult (Rodríguez-Valera et al. 1999).
The sequences obtained from the TSL (TERSS-TSL) and the treatment systems applied to it appeared to be distant from the identified sequences from the database: only 28% of the phylotypes showed more than 97% similarity with previously identified sequences and 31% showed less than 90% similarity. This phenomenon can be explained by the originality of the saline environment screened in this study. Coming from effluent-related samples, it was very likely that the microflora of these ecosystems would be different from the other saline environments that had already been described. In addition, only slightly more than half of the phylotypes were related to already cultured species, which confirms the uncultured majority of halophiles that had already been reported (Litchfield 2004).
Environmental characteristics of tannery effluent-related sludge communities
Among all the environmental parameters considered for statistical analysis, salinity appeared to be the most influential. Hence, the dendrogram and the PCA analysis emphasised the halophilic characteristics of the TSL (TERSS-TSL), the aerobic sequencing batch reactor (TERSS-SBR) and the upflow anaerobic sludge blanket (TERSS-UASB) samples, confirmed by the abundance in these samples of the Gammaproteobacteria and Bacteroidetes that are characteristic of saline environments (Benlloch et al. 2002; Mouné et al. 2003; Venter et al. 2004). A shift in phylum distribution could be observed between TERSS-TSL and the two reactors fed with this influent: TERSS-TSL was dominated by Firmicutes whose proportion was reduced and replaced by Gammaproteobacteria in TERSS-SBR and Bacteroidetes in TERSS-UASB. Regarding TERSS-SBR phylum distribution, it could be stated that the contribution of Proteobacteria and Bacteroidetes (i.e. 64 and 24%, respectively) was similar to that for another aerobic SBR treating hypersaline wastewater generated by tartaric acid production (i.e. 61 and 27% for Proteobacteria and Bacteroidetes, respectively) (Lefebvre et al. 2004).
Regarding TERSS-UASB, the PCA analysis also emphasised its anaerobic characteristics, confirmed by the large number of Firmicutes and Deltaproteobacteria commonly observed in anaerobic digestors (Godon et al. 2004). The population of TERSS-UASB was therefore widely influenced by the double constraint of salt and anaerobiosis, which may explain why sequences from this sample prove to be the most distant from the identified sequences in the database: the communities colonising this type of environment, being at the same time anaerobic and hypersaline, have seldom been described before. Finally, it is worth noting the presence of Synergistes and Thermotogae, neither of which, to our knowledge, have previously been isolated from saline environments.
Finally, the CETP sample (TERSS-CETP), having much lower salinity, was more related to conventional aerobic sludge communities than to saline environments, as can be seen from the dendrogram and the PCA analysis. However, even at this salt content, relatively low in comparison to the other samples, some phylotypes were related to halophilic strains such as Propionibacter pelophilus (AF016690), isolated from estuarine mud and Erythrobacter sp. (AY646157), found in seawater (see Table 2). It can be concluded, therefore, that the selection of halophiles starts at even a low level of salt content, which supports the TERSS-CETP’s position in the PCA analysis, intermediate between-saline environments and conventional aerobic sludge.
Diversity potential of hypersaline wastewater treatment processes
The analysis of the 16S rDNA from whole saline and hypersaline communities has revealed the variable but potentially high diversity of such saline environments (Litchfield 2004). In our study, the saline and hypersaline microbial ecosystems observed by 16S rRNA gene cloning were diverse for the Bacteria lineage. This diversity was estimated by the drawing of rarefaction curves, as well as by the calculation of S ACE. The weakly curvilinear plots for the rarefaction curves obtained for the CETP (TERSS-CETP) and the upflow anaerobic sludge blanket (TERSS-UASB) samples emphasised the need for a higher effort of sampling regarding these samples. On the basis of rarefaction curves, the diversity of TSL (TERSS-TSL) appeared to be higher than that found in some salterns and soda lakes (Benlloch et al. 2002; Humayoun et al. 2003) but lower than that found in seawater and in some other salterns (Mouné et al. 2003; Venter et al. 2004). Such a diversity may surprise at first sight, knowing that salt is used in the tannery process in order to inhibit microbial growth and to preserve the skins from biodegradation. It would seem, therefore, that microbial diversity is not affected by such treatment. The diversity was even higher in the sequencing batch reactor (TERSS-SBR), where the estimated number of phylotypes reached around 100, which is slightly lower than that found in two non-saline aerobic SBR (Bond et al. 1995), and in TERSS-UASB, where the estimated number of phylotypes was estimated to be around 140. The diversity of TERSS-UASB therefore appeared to be similar to that found in a non-saline anaerobic digestor (Godon et al. 1997b). Finally, the last sample analysed in this study consisted of activated sludge collected in a CETP treating the effluents of 128 tanneries. The diversity of TERSS-CETP was higher than that found in the other activated sludge sample (i.e. TERSS-SBR), though comparable to that found in conventional aerobic sludge communities (Bond et al. 1995).
In non-saline wastewater treatment, the efficiency of the process is associated with a high diversity of the microbial community in aerobic (Bond et al. 1995; Snaidr et al. 1997), as well as in anaerobic (Godon et al. 1997b), conditions. In the saline wastewater treatment processes studied in this paper, it has been shown that the efficiency of the process in terms of COD removal was similar to that of the non-saline counterparts (Lefebvre et al. 2005). The diversity found in the hypersaline wastewater (TERSS-TSL) and in the three saline processes (TERSS-SBR, TERSS-UASB and TERSS-CETP) analysed in this paper was also in the same range as that of the non-saline counterpart. Therefore, halophilic sludge should be considered as possessing the same resources of diversity as conventional activated sludge. At this level, the two cannot be distinguished. Consequently, it can be concluded that such halophilic sludge biodiversity enables the biological treatment of hypersaline wastewater to be carried out with an efficiency similar to that observed in the treatment of fresh wastewater, both in aerobic and anaerobic conditions.
References
Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Anton J, Llobet-Brossa E, Rodriguez-Valera F, Amann R (1999) Fluorescence in situ hybridization analysis of the prokaryotic community inhabiting crystallizer ponds. Environ Microbiol 1:517–523
Anton J, Rossello-Mora R, Rodriguez-Valera F, Amann R (2000) Extremely halophilic Bacteria in crystallizer ponds from solar salterns. Appl Environ Microbiol 66:3052–3057
Benlloch S, Martinez-Murcia AJ, Rodriguez-Valera F (1995) Sequencing of bacterial and archaeal 16S rRNA genes directly amplified from a hypersaline environment. Syst Appl Microbiol 18:574–581
Benlloch S, Lopez-Lopez A, Casamayor EO, Ovreas L, Goddard V, Daae FL, Smerdon G, Massana R, Joint I, Thingstad F, Pedros-Alio C, Rodriguez-Valera F (2002) Prokaryotic genetic diversity throughout the salinity gradient of a coastal solar saltern. Environ Microbiol 4:349–360
Bond PL, Hugenholtz P, Keller J, Blackall LL (1995) Bacterial community structures of phosphate-removing and non-phosphate-removing activated sludges from sequencing batch reactors. Appl Environ Microbiol 61:1910–1916
Brosius J, Dull TJ, Sleeter DD, Noller HF (1981) Gene organization and primary structure of a ribosomal RNA operon from Escherichia coli. J Mol Biol 148:107–127
Chao A, Hwang WH, Chen YC, Kuo CY (2000) Estimating the number of shared species in two communities. Stat Sin 10:227–246
Chazdon RL, Colwell RK, Denslow JS, Guariguata MR (1998) Statistical methods for estimating species richness of woody regeneration in primary and secondary rain forests of NE Costa Rica. In: Dallmeier F, Comiskey JA (eds) Forest biodiversity research, monitoring and modeling: conceptual background and Old World case studies. Parthenon Publishing, Paris, pp 285–309
Cole JR, Chai B, Marsh TL, Farris RJ, Wang Q, Kulam SA, Chandra S, McGarrell DM, Schmidt TM, Garrity GM et al (2003) The ribosomal database project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res 31:442–443
Colwell RK (2005) EstimateS: statistical estimation of species richness and shared species from samples. Version 7.5. User’s Guide and application published at: http://www.purl.oclc.org/estimates
Duckworth AW, Grant WD, Jones BE, van Steenbergen R (1996) Phylogenetic diversity of soda lake alkaliphiles. FEMS Microbiol Ecol 19:181–191
Edgcomb VP, Kysela DT, Teske A, de Vera Gomez A, Sogin ML (2002) Benthic eukaryotic diversity in the Guaymas Basin hydrothermal vent environment. Proc Natl Acad Sci USA 99:7658–7662
Godon JJ, Zumstein E, Dabert P, Habouzit F, Moletta R (1997a) Microbial 16S rDNA diversity in an anaerobic digester. Water Sci Technol 36:49–55
Godon JJ, Zumstein E, Dabert P, Habouzit F, Moletta R (1997b) Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl Environ Microbiol 63:2802–22813
Godon JJ, Bayle S, Leclerc M, Delgenes JP (2004) Comparison of 16S ribosomal DNA structure of ten anaerobic digestors. In: Final programm abstracts, 10th Int Symp Microb Ecol. Cancun, Mexico, p 176
Good IL (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40:237–264
Hollibaugh JT, Wong PS, Bano N, Pak SK, Prager EM, Orrego C (2001) Stratification of microbial assemblages in Mono Lake, California, and response to a mixing event. Hydrobiologia 466:45–60
Humayoun SB, Bano N, Hollibaugh JT (2003) Depth distribution of microbial diversity in Mono Lake, a meromictic soda lake in California. Appl Environ Microbiol 69:1030–1042
Jones BE (2004) Industrial enzymes: do halophiles and alkaliphiles have a role to play? In: Ventosa A (ed) Halophilic microorganisms. Springer, Berlin Heidelberg New York, pp 275–284
Lefebvre O, Habouzit F, Bru V, Delgenes JP, Godon JJ, Moletta R (2004) Treatment of hypersaline industrial wastewater by a microbial consortium in a sequencing batch reactor. Environ Technol 25:543–553
Lefebvre O, Vasudevan N, Torrijos M, Thanasekaran K, Moletta R (2005) Halophilic biological treatment of tannery soak liquor in a sequencing batch reactor. Water Res 39:1471–1480
Lefebvre O, Vasudevan N, Torrijos M, Thanasekaran K, Moletta R (2006) Anaerobic digestion of tannery soak liquor with an aerobic post-treatment. Water Res 40:1492–1500
Litchfield CD (2004) Microbial molecular and physiological diversity in hypersaline environments. In: Ventosa A (ed) Halophilic microorganisms. Springer, Berlin Heidelberg New York, pp 49–61
Litchfield CD, Gillevet PM (2002) Microbial diversity and complexity in hypersaline environments: a preliminary assessment. J Ind Microbiol Biotechnol 28:48–55
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, Buchner A, Lai T, Steppi S, Jobb G, Forster W et al (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371
Martinez-Murcia AJ, Acinas SG, Rodriguez-Valera F (1995) Evaluation of prokaryotic diversity by restrictase digestion of 16S rDNA directly amplified from hypersaline environments. FEMS Microbiol Ecol 17:247–255
Mouné S, Caumette P, Matheron R, Willison JC (2003) Molecular sequence analysis of prokaryotic diversity in the anoxic sediments underlying cyanobacterial mats of two hypersaline ponds in Mediterranean salterns. FEMS Microbiol Ecol 44:117–130
Peyton BM, Mormile MR, Alva V, Oie C, Roberto F, Apel WA, Oren A (2004) Biotransformation of toxic organic and inorganic contaminants by halophilic bacteria. In: Ventosa A (ed) Halophilic microorganisms. Springer, Berlin Heidelberg New York, pp 315–331
Quesada E, Bejar V, Ferrer MR, Calvo C, Llamas I, Martinez-Checa F, Arias S, Ruiz-Garcia C, Paez R, Martinez-Canovas MJ, Del Moral A (2004) Moderately halophilic, exopolysaccharide-producing bacteria. In: Ventosa A (ed) Halophilic microorganisms. Springer, Berlin Heidelberg New York, pp 297–314
Rappé MS, Giovannoni SJ (2003) The uncultured microbial majority. Annu Rev Microbiol 57:369–394
Rodríguez-Valera F, Acinas SG, Anton J (1999) Contribution of molecular techniques to the study of microbial diversity in hypersaline environments. In: Oren A (ed) Microbiology and biogeochemistry of hypersaline environments. CRC Press, Boca Raton, pp 27–38
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sekiguchi Y, Kamagata Y, Syutsubo K, Ohashi A, Harada H, Nakamura K (1998) Phylogenetic diversity of mesophilic and thermophilic granular sludges determined by 16S rRNA gene analysis. Microbiology 144:2655–2665
Snaidr J, Amann R, Huber I, Ludwig W, Schleifer KH (1997) Phylogenetic analysis and in situ identification of bacteria in activated sludge. Appl Environ Microbiol 63:2884–2896
Snell-Castro R, Godon J-J, Delgenes J-P, Dabert P (2005) Characterisation of the microbial diversity in a pig manure storage pit using small subunit rDNA sequence analysis. FEMS Microbiol Ecol 52:229–242
Sorokin DY, Kuenen JG (2005) Haloalkaliphilic sulfur-oxidizing bacteria in soda lakes. FEMS Microbiol Rev 29:685–702
Sreeram KJ, Ramasami T (2003) Sustaining tanning process through conservation, recovery and better utilization of chromium. Resour Conserv Recycling 38:185–212
Stoop MLM (2003) Water management of production systems optimised by environmentally oriented integral chain management: case study of leather manufacturing in developing countries. Technovation 23:265–278
Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu DY, Paulsen I, Nelson KE, Nelson W, Fouts DE, Levy S, Knap AH, Lomas MW, Nealson K, White O, Peterson J, Hoffman J, Parsons R, Baden-Tillson H, Pfannkoch C, Rogers YH, Smith HO (2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science 304:66–74
Ward BB, Martino DP, Diaz MC, Joye SB (2000) Analysis of ammonia-oxidizing bacteria from hypersaline Mono Lake, California, on the basis of 16S rRNA sequences. Appl Environ Microbiol 66:2873–2881
Wiegant WM, Kalker TJJ, Sontakke VN, Zwaag RR (1999) Full scale experience with tannery water management: an integrated approach. Water Sci Technol 39:169–176
Acknowledgements
This work was supported by the Indo-French Cell for Bioprocesses on Environment (IFCBE) based on cooperation between the Centre for Environmental Studies, Anna University, Chennai (India) and the French National Institute for Agricultural Research (Institut National de la Recherche Agronomique, INRA). The authors would like to thank Dr. P. Dabert and Ms. V. Bru for their assistance during the study and Dr. M. Moletta for providing data regarding anaerobic sludge diversity.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by K. Horikoshi
Rights and permissions
About this article
Cite this article
Lefebvre, O., Vasudevan, N., Thanasekaran, K. et al. Microbial diversity in hypersaline wastewater: the example of tanneries. Extremophiles 10, 505–513 (2006). https://doi.org/10.1007/s00792-006-0524-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-006-0524-1