
Abstract
The ability of a psychrotolerant microbial consortium to degrade crude oil at low temperatures was investigated. The enriched arctic microbial community was also tested for its ability to utilize various hydrocarbons, such as long-chain alkanes (n-C24 to n-C34), pristane, (methyl-)naphthalenes, and xylenes, as sole carbon and energy sources. Except for o-xylene and methylnaphthalenes, all tested compounds were metabolized under conditions that are typical for contaminated marine liquid sites, namely at pH 6–9 and at 4–27°C. By applying molecular biological techniques (16S rDNA sequencing, DGGE) nine strains could be identified in the consortium. Five of these strains could be isolated in pure cultures. The involved strains were closely related to the following genera: Pseudoalteromonas (two species), Pseudomonas (two species), Shewanella (two species), Marinobacter (one species), Psychrobacter (one species), and Agreia (one species). Interestingly, the five isolated strains in different combinations were unable to degrade crude oil or its components significantly, indicating the importance of the four unculturable microorganisms in the degradation of single or of complex mixtures of hydrocarbons. The obtained mixed culture showed obvious advantages including stability of the consortium, wide range adaptability for crude oil degradation, and strong degradation ability of crude oil.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrocarbons are the major pollutants in marine environments. They are derived from terrestrial and freshwater run-off, offshore oil production, refuse from coastal oil refineries, shipping activities, and accidental spillage of fuels and other petroleum products. Although the majority of hydrocarbon-derived contaminations occur in cold marine environments, most of the investigations have been performed at higher temperatures, namely between 20 and 35°C (Raghukumar et al. 2001; Ko et al. 1999; Geerdink 1997; Margesin and Schinner 1997a, b; Rosenberg et al. 1992). For a long time, decreased solubility was considered to be responsible for the recalcitrance of hydrophobic compounds observed in temperate and cold environments. Several recent reports, however, have indicated that some bacteria may have adapted to the low solubility of hydrophobic environmental chemicals and that generalizations about the bioavailability of hydrocarbons might be inappropriate (Wick et al. 2002a; Bastiaens et al. 2000; Friedrich et al. 2000; Grosser et al. 2000; Guerin and Boyd 1992). Indeed, hydrocarbons are degraded at rates which exceed their rates of dissolution in the aqueous phase, demanding special uptake mechanisms to be employed by hydrocarbon-degrading microorganisms (Leahy and Colwell 1990; Thomas et al. 1986).
In cold climates, psychrophilic and psychrotolerant microorganisms play an important role in the biodegradation of organic matter. These bacteria are distinguished by their minimum, optimum, and maximum growth temperatures, which are <0, <15, and <20°C, for psychrophilic and 0–5, >15, and >20°C for psychrotolerant bacteria (Morita 1975). Former studies concerning degradation of hydrocarbons by psychrotolerant bacteria were performed with single bacterial strains (Whyte et al. 1998; Geerdink et al. 1996) and in the presence of complex compounds such as yeast extract (Hamme van et al. 2000). The obtained cell yields under these conditions in most cases do not correspond to the degradation of hydrocarbons. Furthermore, during subcultivation of the strains plasmids, which encode enzymes with essential degradation capabilities, can be lost. Former investigations have shown that linear and in some cases branched alkanes from crude oil or diesel fuel are degradable while aromatic compounds were hardly attacked (Raghukumar et al. 2001; Whyte et al. 1998; Atlas and Bartha 1992). These studies indicate that pure cultures can metabolize only a limited range of hydrocarbons. Consequently, mixed populations with overall broad enzymatic capacities are required to degrade complex mixtures of hydrocarbons such as crude oil or diesel fuel (Leahy and Colwell 1990). Such mixed cultures display metabolic versatility and superiority to pure cultures (Hamme et al. 2000). In a consortium, single strains can complement each other, e.g. by co-metabolic turnover reactions or by interactions with substrates (e.g. via biofilm formation) and carry out finally a more effective degradation process. Pelz et al. (1999) have proved for example that in a consortium metabolic and physiological weaknesses of primary degraders of hydrocarbons can effectively be compensated by recruitment of other organisms from the mixed culture with appropriate complementary physiology.
Based on the fact that most hydrocarbon-derived pollutions occur at low temperatures, attempts were made to enrich a microbial consortium effective in crude oil degradation in cold habitats. For these studies, physiological experiments and molecular biological techniques were employed to analyze the microbial community in the consortium.
Materials and methods
Source of bacteria
During an expedition to Spitzbergen in 1998, arctic sea ice and seawater samples were collected at Svalbard (Norway) and transported to the laboratory at a temperature of 2–10°C. Samples from different habitats were mixed and used as inoculum for the enrichment culture.
Crude oils
Two different crude oils (nos. 1 and 2) were used for degradation experiments. Both consisted of linear and branched alkanes (pristane and phytane) and diverse aromatic components (e.g. toluene, xylenes, ethylbenzene, (methylated) naphthalenes, dibenzothiophenes, phenanthrenes, and acenaphthenes) in varying concentrations. The oils were derived from different offshore North Sea oil fields. Unless otherwise stated in the text, oil no. 1 was used.
Media
The enrichment medium contained (g l−1) NaCl 28.13; KCl 0.77; CaCl2 × 2 H2O 0.02; MgSO4 × 7 H2O 0.5; NH4Cl 1.0; FeCl2 0.001; yeast extract 0.5. The pH was adjusted to 7.0. After autoclaving the medium, the following sterile solutions were added: 1 ml of tenfold trace element solution as described for medium 141 (DSMZ 1998); 100 ml containing KH2PO4 (2.3 g) and Na2HPO4 × 2 H2O (2.9 g) (pH 7.2); 100 ml containing Na-acetate (0.5 g), Na-succinate (0.5 g), DL-malate (0.5 g), Na-pyruvate (0.5 g), D-mannitol (0.5 g), and glucose (2.0 g) (pH 6.8), to give a total volume of 1000 ml. For preparation of a mineral medium, the carbon sources and yeast extract were omitted. Isolation of strains was performed on TSA agar plates (Sifin, Berlin, Germany).
Enrichment and cultivation
Enrichment was performed in a one-liter flask containing 500 ml of enrichment medium with 10 ml of crude oil. As inoculum, 10 ml of the sample from the arctic sea ice was applied. Incubation was performed at 4°C for 6 weeks without regular shaking, as described by Mohn et al. (1997). To avoid anaerobic conditions, the flask was mixed and aerated manually each third day. Microbial growth was monitored by direct cell counting using a Neubauer counting chamber. After significant cell growth had been achieved, the bacteria were subcultivated in 100-ml Erlenmeyer flasks (triplicates) containing 22.5 ml of minimal medium (final concentration: 4–5 × 107 cells ml−1). Crude oil was added as a single source of carbon and energy (1 ml l−1). The flasks were sealed tightly with screw caps (Whyte et al. 1998) containing PTFE covered with silicone septa to avoid abiotic loss of the substrate. After initial growth was obvious, the cultures were aerated once a day and mixed thoroughly. Uninoculated control flasks (duplicates) were incubated and aerated in parallel to monitor abiotic losses of the substrates. In addition, the following carbon sources were used in the growth experiments: 1 mM naphthalene, methylnaphthalenes, n-tetracosane (n-C24), n-tetratriacontane (n-C34), 0.5 mM pristane, and 0.1 mM xylenes.
Gas chromatography
For chemical analysis, samples with a volume of 23.5 ml were extracted with 5 ml hexane, dried over Na2SO4, and stored at 4°C in 2-ml glass vials sealed with screw caps. Gas chromatographical measurement of the hexane soluble fraction of the crude oil during the degradation experiments was accomplished using a Perkin Elmer Autosystem Gas Chromatograph (GC), (Überlingen, Germany) fitted with a flame ionization detector (FID). The used column was a 30 m × 0.32 mm (ID) glass capillary column Rtx-5 (Restek, Bad Soden, Germany) with a film thickness of 0.25 μm. Hydrogen was used as the carrier gas. Injector mode was performed with a split volume of 90 ml, at 300°C; injection volume was 2 μl. The oven program started at 35°C, held for 2 min, and was raised at 4°C min−1 to 310°C, and held for 10 min. Detector temperature was adjusted to 300°C. Phenanthrene- d10 was used as internal standard (15 mg/l); 2 μl was added to 50 μl of the sample.
For calculation of total crude oil degradation, peak areas (from triplicate samples) were quantified by integration. These values were expressed as the percentage degraded relative to the amount of the corresponding components, which remained in the appropriate abiotic control samples.
Mass spectrometry
Analysis with gas chromatograph-mass spectrometer (GC-MS) was performed as described by Annweiler et al. (2000). The measurements were carried out with a Carlo Erba 4160 GC, using a DB-5 HT capillary column (J & W Scientific, Folsom, USA) with a length of 60 m; (ID 0.32 mm) and a film thickness of 0.25 μm. Carrier Gas and temperature program were applied as described for the GC measurements. The GC was coupled with a CH7A mass spectrometer (Finnigan, Germany). The conditions were as follows: ionization mode, EL+; ionization energy, 70 eV; emission current, 200 μA; temperature of source 250°C; mass range m/z 50–500.
16S rDNA amplificaton and cloning
Cells were harvested from 2-ml cultures by centrifugation and resuspended in 100 μl of minimal medium. An aliquot of 1 μl was used as a template for amplification of 16S rRNA genes with 27F and 1492R primers (Lane 1993). The PCR was performed in a total volume of 100 μl containing 50 pmol of each primer, 200 μM of each dNTP, 30 μg bovine serum albumin, 20 mM Tris–HCl, 50 mM KCl, and 1.5 mM MgCl2. Negative controls without DNA template were included in every reaction set. After applying a hot start (Muyzer et al. 1993) with 2.5 U Taq DNA polymerase (GibcoBRL Life technologies, Karlsruhe, Germany), 30 cycles of 94°C for 1.5 min, 46°C for 1.5 min, and 72°C for 1.5 min were run in a DNA thermal cycler Gene Amp PCR System 2400 (Perkin–Elmer, Überlingen, Germany).
The amplicons were separated on 1.0% agarose gel stained with ethidium bromide and purified with the QIAquick purification Kit (Qiagen, Hilden, Germany) or with the Qiaex Gel extraction Kit (Qiagen). Purified 16S rDNA fragments were cloned by the TA-TOPO cloning kit (Invitrogen, Karlsruhe, Germany) in pCR2.1-TOPO vector following the manufacturer’s instructions. For DGGE and sequencing, plasmids were isolated and purified with a QIAprep Spin Miniprep Kit (Qiagen).
Denaturing gradient gel electrophoresis (DGGE)
Primers GM5clamp and 907R were used to amplify variable regions of the 16S rDNA in a touchdown PCR, as described by Sahm et al. (1999) and by Buchholz-Cleven et al. (1997) with the following modifications: as template served 0.5–1.0 μg of DNA extract, prepared from 2 ml bacterial culture with the QIAamp DNA Mini Kit (Qiagen). For reamplification of PCR bands, DNA was excised from the gel and eluted overnight in 100 μl H2O MilliQ at 4°C. For the new amplification, 1 μl of eluted DNA served as the template.
The hot start was followed by 25 cycles at 94°C for 1.0 min, 62°C for 1.0 min, and 72°C for 3.0 min. The annealing temperature was decreased by 0.4°C in every cycle until a touchdown at 52°C was achieved. From cycle 11 to 25, the elongation time was increased (0.4 min). Additionally, five cycles were carried out at 94°C for 1.5 min, 52°C for 1.5 min, and 72°C for 7.4 min. The DGGE was performed with a DGGE Biorad DCodeTM System (Biorad Laboratories Inc., München, Germany) as described previously (Muyzer et al. 1996).
The gels contained denaturing gradients ranging from 20 to 80%. A 150 ml of 80% denaturing acrylamide solution contained 22.5 ml acrylamide (40%, 37.5:1, Biorad Laboratories Inc, München, Germany), 3 ml 50 × TAE, 48 ml formamide (deionized, Roth, Germany), and 50.4 g urea. This solution was stored in a dark bottle. A 0% denaturing acrylamide solution contained the same ingredients except for urea and formamide. Electrophoresis was run in 1 × TAE at 55°C for 5 or 20 h at 200 or 100 V, respectively.
Sequencing and phylogenetical analysis
The Taq DyeDesoxy Terminator Cycle Sequencing kit (Applied Biosystems) was used to directly sequence the purified PCR products. Sequencing reactions were analyzed on the 373S DNA sequencer (Applied Biosystems). Both strands of the amplification product were sequenced using primers 27F, 787F, 787R, 1175R, 1099F, and 1492R (Lane 1993). Sequences were compared to the available 16S rDNA primary structures present in the EMBL database by using the Fasta3 program. To detect chimera, the chimera_check program at RDPII was used.
Results
Enrichment of a crude oil degrading consortium
A crude oil degrading bacterial consortium was enriched at 4°C from a mixture of arctic sea ice and seawater samples derived from Spitzbergen. For enrichment and cultivation, the cultures were not permanently shaken because continuous shaking at 120 rpm inhibited microbial growth significantly (data not shown). Microscopic examination revealed the oil droplets to be covered by a biofilm before effective degradation was observed (Fig. 1a). After approximately 19 days of cultivation at 4°C, the size of the oil droplets decreased significantly (Fig. 1b) and finally the droplets disappeared. The center of the droplets was not occupied by microorganisms. Cells in the vicinity of the oil droplets were found to be very motile and did not form any aggregates. By applying gas chromatography, the degradation of saturated hydrocarbons in crude oil was analyzed. During the degradation process, n-alkanes (C8–C34) and the isoprenoids pristane and phytane were found to be completely eliminated after 28 days of incubation at 4°C (Fig. 2). Short-chain n-alkanes (C8–C14) were preferentially metabolized by the consortium. These components were used during the first 2 weeks. Afterwards, n-alkanes with a chain length of 15–34 carbon atoms were attacked in parallel with the isoprenoids pristane and phytane.

Growth of the psychrotolerant consortium on crude oil (0.1%) at 4°C for 6 days (a) and 19 days (b). Biofilm formation after cultivation of the consortium at 4°C on n-C34 crystals (1 mM) for 4 weeks (c). Experiments were performed in a mineral medium at pH 7.0, scale bar 10 μm

GC profiles of crude oil extracted from the aqueous phase after cultivation of the psychrotolerant consortium in a mineral medium with crude oil at 4°C. a abiotic control (28 days); b inoculated sample after 14 days of growth; c inoculated sample after 28 days. IS internal standard (phenanthrene d10), 8–34, n-alkanes (numbers designate the number of C atoms), pr pristine, ph phytane. Alkanes, pristine, and phytane were identified by comparison of the retention time and mass spectra with authentic standards
Compared to abiotic controls, mass spectrometric analysis provided a clear indication of the degradation of diverse aromatic components in crude oil at 4°C (Fig. 3). Ethylbenzene, m-xylene, and p-xylene, e.g. were found to be utilized by the consortium (Fig. 3a). In contrast, o-xylene was not attacked. A temporary relative enrichment of p-xylene and ethylbenzene during crude oil degradation indicated a preferential use of m-xylene (Fig. 3a). Furthermore, several alkyl substituted benzenes with nine and ten carbon atoms and naphthalene were totally eliminated by the consortium (Fig. 3b–d), whereas other isomers remained unattacked. Even 2-methylnaphthalene was attacked during the degradation process, indicated by the relative enrichment of 1-methylnaphthalene. Total elimination of these two isomers, however, could not be observed (Fig. 3d). In contrast, other polyaromatic hydrocarbons (e.g. phenanthrene, dibenzothiophene, and acenaphthene) and saturated cyclic compounds (hopanes) were not attacked by the consortium even after 5 weeks of incubation at 4°C. In conclusion, the conversion of hydrophobic substrates like long-chain n-alkanes and naphthalene is astonishing, especially at 4°C. It can be estimated that the metabolism of these components occurs at rates which exceed their rates of dissolution in the medium.

Mass chromatograms of aromatic components of crude oil during microbial degradation at 4°C; first line from the top abiotic control after 28 days, second line inoculated sample after 7 days, third line inoculated sample after 28 days. a xylenes and ethylbenzene (ion chromatogram m/z 106), b alkylsubstituate benzenes with nine carbon atoms (ion chromatogram m/z 120), c alkylsubstituate benzenes with ten carbon atoms (ion chromatogram m/z 132), d naphthalene, and methylnaphthalenes (ion chromatograms m/z 128+144). o o-xylene, m m-xylene, p p-xylene, e ethylbenzene, n naphthalene, 1-m 1-methylnaphthalene, 2-m 2-methylnaphthalene, peaks in a and d were identified by coinjection of standards. Arrows indicate decreasing/disappearing peaks
Further physiological experiments were performed to find the optimal conditions for crude oil degradation. Bacterial degradation of crude oil was observed at neutral pH (7.0) in a temperature range between 4 and 27°C, with an optimum temperature of 20°C. The corresponding growth rates were found to be nearly identical (0.83–0.9 day−1) in a temperature range from 15 to 27°C (Fig. 4). The degradation process was completed within 7 days at 20°C and within 28 days at 4°C. No degradation was observed at 30°C. Furthermore, crude oil degradation was observed in a pH range between 6 and 9 with an optimum at pH 8.5 (data not shown). At pH 8.5, up to 77% of the crude oil was degraded at 20°C within 7 days and up to 70% within 4 weeks at 4°C. The degradation pattern was reproducible when a different crude oil was used as substrate (oil no. 2). In this case, 65% degradation was achieved at 4°C after 5 weeks and 71% at 20°C after 10 days (data not shown). From these results, it is obvious that the metabolic capability of the consortium is not restricted to one type of crude oil.

Effect of temperature on growth of the consortium on crude oil. Growth rates of the psychrotolerant mixed culture were determined by cell counting in exponential growth phases at different temperatures with crude oil as sole carbon source in a mineral medium at pH 7.0. The increase in cell growth was correlated with the decrease in crude oil concentration (data not shown)
Utilization of typical crude oil components
To analyze whether the decrease in oil components is due to mineralization or co-metabolic turnover reactions, various crude oil components (n-C24, n-C34, pristane, xylenes, and (methyl-)naphthalene) were tested as single sources of carbon and energy for growth. Incubation was performed at 4 and 20°C in the mineral medium at neutral pH (7.0). All substrates except o-xylene and methylnaphthalenes supported the growth of the consortium. Interestingly, hydrophobic n-alkane crystals (C24 and C34) were covered by a biofilm, when applied as a single carbon source (Fig. 1c). The n-C24 crystals disappeared from the medium and the size of n-C34 crystals was significantly reduced. The growth rates for n-C24 at 4 and 20°C were 0.32 (day−1) and 0.4 (day–1), respectively. Corresponding values for n-C34 were found to be 0.19 (day–1) at 4°C and 0.29 (day−1) at 20°C. As already described for crude oil, pristane droplets were covered at 4°C by a biofilm, before they were utilized. The growth rates at 4 and 20°C were 0.31 (day−1) and 0.72 (day−1), respectively. From the three-xylene isomers, only m-xylene and p-xylene enabled microbial growth. Selective elimination of these two components from crude oil was also shown by mass spectrometry. During growth, biofilm formation could be observed on the surface of the medium as well. The corresponding growth rates were 0.15 (day−1) at 4°C and 0.38 (day−1) at 20°C for m-xylene and 0.21 (day−1) at 4°C and 0.51 (day−1) at 20°C for p-xylene. To a lower extent, biofilm formation was observed during the growth of the consortium on naphthalene. Thus, growth rates for this substrate were only 0.15 (day−1) at 4°C and 0.22 (day−1) at 20°C. In spite of the low growth rates, naphthalene crystals were eliminated from the medium. This was, however, accompanied by the formation of a yellow-brown-colored metabolite. Further growth experiments with 1-methylnaphthalene and 2-methylnaphthalene as sole carbon sources have shown that these substrates did not support growth even after 5 weeks of cultivation at various temperatures. These observations are in accordance with results obtained from GC–MS analysis, where only a slight decrease in 2-methylnaphthalene could be observed.
Phylogenetical analysis
To analyze the composition of the consortium, classical and molecular biological methods were used. At first, the culturable strains were separated from each other by streaking several times on TSA-agar plates. With this method, five bacterial isolates were obtained, which were described as P1, P3, P4, P5, and P6. Based on 16S rDNA sequences, the isolates were closely related to the following strains: P1, Pseudoalteromonas elyakovii (99.6%); P3, Psychrobacter glacincola (98.9%); P4, Pseudomonas anguilliseptica (97.8%); P5, Pseudomonas synxantha (95.3%); P6, Agreia bicolorata (96.6%).
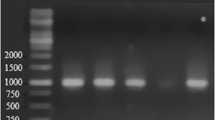
To identify the unculturable strains, 16S rDNA fragments derived from the consortium were cloned. Afterwards, DGGE fragments from the mixed culture and from the clones were amplified and analyzed by a corresponding gel (Fig. 5). Interestingly, variations in the band patterns of the mixed culture were observed depending on the growth phase. Only the samples from exponential and stationary phase together were able to display the full bacterial diversity of the investigated mixed culture (Fig. 5, lanes 1, 4, 7, 10, and 13). After the screening of 117 clones, all DGGE bands of the consortium could be assigned, either to DGGE fragments from the isolated strains or to corresponding DNA from selected clones (C51; C53; C56; C81), (Fig. 5).

Analysis of the microbial population by DGGE. lanes 1, 4, and 7 show the band pattern of a sample taken in the stationary phase of growth after crude oil degradation at 4°C. The lanes 10 and 13 show the corresponding band patterns of the consortium at the exponential growth phase. The remaining lanes belong to the following isolates or clones: lane 2 isolate P3, lane 3 isolate P5, lane 5 clone 53, lane 6 isolate P1, lane 8 clone 81, lane 9 clone 51, lane 11 isolate P4, lane 12 clone 56, lane 14 isolate P6
Due to the fact that each step in PCR-mediated community analysis is potentially open to error or bias, the clones 51, 53, 56, and 81 were checked for the existence of chimeric sequences. Clone 51 was a hybrid, assembled under the conditions used in the PCR reaction. The clone displayed 94.5% similarity to Marinobacter hydrocarbonoclasticus over the first 1150 nucleotides and a similarity of 99.6% to P. elyakovii in the last 250 nucleotides. Obviously, the second part of the clone belongs to isolate P1. This strain displays the highest similarity to P. elyakovii (see above) over a sequence length of 1420 bp. Consequently, the DGGE band from the consortium, which corresponded to clone 51 was excised in duplicate and sequenced as well. Over a length of 550 bp, it displayed a similarity of 93.8% to M. hydrocarbonoclasticus, indicating that indeed a Marinobacter strain might be present in the community. On the level of 16S rDNA sequences, the clones C53, C56, and C81 displayed highest similarities to Pseudoalteromonas atlantica (99.2%), Shewanella baltica (97.4%), and Shewanella frigidimarina (99.9%). However, none of these clones contained chimeric sequences.
Degradation capabilities of the pure strains and the defined mixed cultures
To analyze the potential role of culturable strains in the described degradation processes, all isolates (P1, P3, P4, P5, and P6) were tested separately for their ability to degrade crude oil components (n-alkanes (C24 and C34), pristane, xylenes, naphthalene, methyl-naphthalenes). None of the five isolated strains showed significant growth with any of the mentioned substrates compared to the original consortium; biofilm formation could neither be observed.
Furthermore, the same strains were cultured separately and in defined mixed cultures with crude oil as the sole carbon source. The defined mixed cultures contained 2–5 isolates in various combinations. Only isolate P3 adhered to crude oil droplets and formed a biofilm when cultivated alone with the carbon source. No significant degradation of the substrate, however, was observed. These experiments clearly show that none of the culturable strains (separately or in combination) are effective in crude oil degradation (data not shown). From these experiments, it is obvious that the nonculturable strains are essential and play a key role in the degradation of crude oil.
Discussion
In contrast to earlier reports where psychrophilic or mesophilic single strains were used (Whyte 1998; Atlas and Bartha 1992; Raghukumar et al. 2001), the investigated consortium presented in this study is able to degrade not only linear alkanes but also isoprenoids (pristane and phytane) and several aromatic compounds (ethylbenzene, m-xylene/p-xylene, naphthalene). The consortium was able to co-metabolize isoprenoids with long-chain n-alkanes. In previous reports, isoprenoid metabolism was only observed after the complete elimination of the linear alkanes (Whyte et al. 1998; Atlas and Bartha 1992; Geerdink et al. 1996; Leahy and Colwell 1990; Westlake et al. 1974; Jobsen et al. 1972). In this study, C8–C15 n-alkanes were preferably metabolized followed by C16–C36 n-alkanes. This sequential degradation of n-alkanes was already reported by Peters and Moldowan (1993) and is also in agreement with previous investigations performed at low temperatures (Whyte et al. 1998), indicating that the solubility of individual components influences their bioavailability even at low temperatures.
The utilization of the diaromatic compound naphthalene by the consortium at 4°C, at least via co-metabolism, is remarkable. Leahy and Colwell (1990) reported on the degradation of asphaltenic compounds in mixed bacterial cultures to be dependent upon the presence of n-alkanes (12–18 carbon atoms in length). Also, the induction of enzymes for PAH degradation can depend on the presence of lower-molecular-weight aromatics such as naphthalene (Atlas and Bartha 1992). Saturated, cyclic high-molecular-weight compounds like hopanes were not degraded by the investigated consortium. Hopenes belong to the most persistent components of oil spillages in the environment. From the light aromatic crude oil fraction, toluene and o-xylene were not attacked by the consortium. The presence of these components obviously did not inhibit crude oil degradation. Bacteria that degrade xylenes commonly fall into two classes: those that can degrade both m-xylene and p-xylene, and those that can degrade o-xylene only. It can be concluded that the metabolic pathway for utilization of o-xylene is missing in the consortium.
It is generally believed that bacteria could utilize only solubilized hydrocarbons (Britton 1984). Our results with the crystalline substrates (n-C24, n-C34, and naphthalene) indicate that the general view about substrate bioavailability might be inappropriate (Wick et al. 2002a; Bastiens et al. 2000; Friedrich et al. 2000; Grosser et al. 2000; Guerin and Boyd 1992). The bioavailability of hydrophobic compounds can be controlled by factors other than solubility because degradation of these substrates occurs at rates which exceed the rates of hydrocarbon dissolution (Jobson et al. 1972). It is obvious that microorganisms have developed various strategies, which enable them to utilize insoluble hydrocarbons efficiently. Biofilm formation, which was observed in most cases, could be applied as a strategy for the utilization of crystalline components (Bowman et al. 1997; Whyte et al. 1999; Wick et al. 2001, 2002a, b; Watkinson and Morgan 1990).
Although the microbial population was enriched at 4°C, the optimal temperature for growth and crude oil degradation was 20°C. Psychrotolerant bacteria usually possess an optimum temperature 10–20°C above the environmental temperatures from which they were isolated (Morita 1975). The culturable strains were closely related to P. elyakovii (99.6%), Psychrobacter glacincola (98.9%), P. anguilliseptica (97.8%), P. synxantha (95.3%), and A. bicolorata (96.6%). None of these strains, however, were previously reported to degrade crude oil. Interestingly, the DGGE band profile of the consortium was found to be dependent on the growth. Similar variations in bacterial profiles of mixed cultures were reported by Ferris et al. (1996) and Gillan et al. (1998). Furthermore, the detection limit for a bacterial member in a consortium was found to be <1% by applying DGGE (Polz and Cavanaugh 1998). Although the composition of microbial communities determined by the analysis of libraries of cloned PCR-amplified sequences is not quantitative (von Witzingerode et al. 1997), the predominance of sequence types suggests that the microorganisms represented by these sequences play an important role in the microbial activity within the psychrotolerant consortium.
The closest relative to the unculturable strain C51, M. hydrocarbonoclasticus, was isolated from seawater near a petroleum refinery (Gauthier et al. 1992). The strain displays a high tolerance against NaCl, up to 20% (Gauthier et al. 1992). Moreover, the reference strain M. hydrocarbonoclasticus utilizes linear saturated hydrocarbons (tetradecane, hexadecane, eicosane, and heneicosane) with high degradation rates. Growth to a lower extent was also observed with pristane, phenyldecane, and phenanthrene as carbon and energy sources (Gauthier et al. 1992). In general, M. hydrocarbonoclasticus belongs to the highly specialized hydrocarbonoclastic marine bacteria, which are present only in low numbers in “clean” marine environments but “bloom” (may account for up to 90% of microbial community) in response to oil spills or other hydrocarbon contamination events (Yakimov et al. 2002; Kasai et al. 2001; Syutsubo et al. 2001; Harayama et al. 1999). The close genetic relationship to M. hydrocarbonoclasticus suggests that the bacterial strain, corresponding to clone C51, may provide physiological capabilities to degrade crude oil components with respect to the overall degradation potential of the consortium.
Interestingly, a close relationship between C53 and P. atlantica was found. This strain exhibits the ability to switch the production of the adhesion EPS on and off. By this process, the bacterium moves from solid surfaces (such as seaweed or sand) to the Open Ocean and then back to a solid substrate to initiate biofilm formation (Corpe 1970). A 1.2-kb multicopy insertion sequence was found to be responsible for this phenomenon (Bartlett et al. 1988; Belas and Silverman 1989). The close relationship of C53 to P. atlantica may indicate that C53 could play an essential role in biofilm formation prior to the described degradation processes by the consortium. S. baltica, the closest related strain of C56 was originally isolated from an oil brine (Ziemke et al. 1998) but until now it has not been reported to degrade pollutants of environmental relevance.
References
Al-Mallah M, Gout M, Mille G, Bertrand JC (1990) Production of emulsifying agents during growth of a marine Alteromonas in sea water with eicosane as carbon source, a solid hydrocarbon. Oil Chem Pollut 6:289–305
Annweiler E, Richnow HH, Antranikian G, Hebenbrock S, Harms C, Franke S, Franke W, Michaelis W (2000) Naphthalene degradation and incorporation of naphthalene-derived carbon into biomass by the thermophile Bacillus thermoleovorans. Appl Environ Microbiol 66:518–523
Atlas MA, Bartha R (1992) Hydrocarbon biodegradation and oil spill bioremediation. In: Marshall KC (ed) Advances in microbial ecology, vol 12. Plenum Press, New York, pp 287–338
Bartlett DH, Wright ME, Silverman M (1988) Variable expression of extracellular polysaccharide in the marine bacterium Pseudomonas atlantica is controlled by genome rearrangement. Proc Natl Acad Sci USA 85:3923–3927
Bastiens L, Springael D, Wattiau P, Harms H, de Wachter R, Verachtert H, Diels L (2000) Isolation of new polycyclic aromatic hydrocarbon (PAH) degrading bacteria using PAH sorbing carriers. Appl Environ Microbiol 66:1834–1843
Belas DH, Silverman M (1989) Nucleotide sequence of IS492, a novel insertion sequence causing variation in extracellular polysaccharide production in the marine bacterium Pseudomonas atlantica. J Bacteriol 171:1763–1766
Bowman JP, McCammon SA, Nichols DS, Skerratt JH, Rea SM, Nichols PD, McMeekin TA (1997) Shewanella gelidimarina sp. nov. and S. frigidimarina sp. nov., novel antarctic species with the ability to produce eicosapentaenoic acid (20:5ω3) and grow anaerobically by dissimilatory Fe (III) reduction. Int J Syst Bacteriol 47:1040–1047
Buchholz-Cleven BE, Rattunde EB, Straub KL (1997) Screening of genetic diversity of isolates of anaerobic Fe(II)-oxidizing bacteria using DGGE and whole-cell hybridization. Syst Appl Microbiol 20:301–309
Corpe WA (1970) Adhesion of marine bacteria to solid surfaces. In: Manley RS (ed) Adhesion in biological systems. Academic Press, New York, pp 73–87
Ferris MJ, Muyzer G, Ward DM (1996) Denaturing gradient gel electrophoresis profiles of 16S rRNA-defined population inhabiting a hot spring microbial mat community. Appl Environ Microbiol 62:340–346
Friedrich M, Grosser RJ, Kern A, Inskeep WP, Ward DM (2000) Effect of model sorptive phases on phenanthrene degradation: molecular analysis of enrichments and isolates suggests selection based on bioavailability. Appl Microbiol Biotechnol 66:2703–2710
Gauthier MJ, Lafay B, Christen R, Fernandez L, Acquaviva M, Bonin P, Bertrand JC (1992) Marinobacter hydrocarbonocalsticus gen. nov., sp. nov., a new, extremely halotolerant, hydrocarbon-degrading marine bacterium. Int J Syst Bacteriol 42:568–576
Geerdink MJ, van Loosdrecht MCM, Luyben KChAM (1996) Biodegradability of diesel oil. Biodegradation 7:73–81
Gillan DC, Speksnijder AGCL, Zwart G, De Ridder C (1998) Genetic diversity of the biofilm covering Montacuta ferruginose (Mollusca, bivalvia) as evaluated by denaturing gradient gel electrophoresis analysis and cloning of PCR-amplified gene fragments coding for 16S rRNA. Appl Environ Microbiol 64:3464–3472
Grosser RJ, Friedrich M, Ward DM, Inskeep WP (2000) Influence of different chemical treatments on transport of Alcaligenes paradoxus in porous media. Appl Environ Microbiol 61:1750–1756
Guerin WF, Boyd SA (1992) Differential bioavailability of soil-sorbed naphthalene to two bacterial species. Appl Environ Microbiol 58:1141–1152
Hamme van JD, Odumeru JA, Ward OP (2000) Community dynamics of a mixed-bacterial culture growing on petroleum hydrocarbons in batch culture. Can J Microbiol 46:441–450
Harayama S, Kishira H, Kasai Y, Shutsubo K (1999) Petroleum biodegradation in marine environments. J Mol Microbiol Biotechnol 1:63–70
Jobson A, Cook FD, Westlake DWS (1972) Microbial utilization of crude oil. Appl Microbiol 23:1082–1089
Kasai Y, Kishira H, Syutsubo K, Harayama S (2001) Molecular detection of marine bacterial populations on beaches contaminated by the Nakhodka tanker oil-spill accident. Environ Microbiol 3:246–255
Ko SH, Lebault JM (1999) Effect of a mixed culture on co-oxidation during the degradation of saturated hydrocarbon mixture. J Appl Microbiol 87:72–79
Lane DJ (1993) In: Goodfellow M, O’Donnell AG (eds) Handbook of new bacterial systematics. Academic Press, London, pp 115–194
Leahy JG, Colwell RR (1990) Microbial degradation of hydrocarbons in the environment. Appl Environ Microbiol 54:305–315
Margesin R, Schinner F (1997a) Bioremediation of diesel-oil-contaminated alpine soils at low temperatures. Appl Microbiol Biotechnol 47:462–468
Margesin R, Schinner F (1997b) Efficiency of indigenous and inoculated Cold-adapted soil microorganisms for biodegradation of diesel oil in saline soils. Appl Environ Microbiol 63:2660–2664
Mohn WW, Westerberg K, Cullen WR, Reimer KJ (1997) Aerobic biodegradation of biphenyl and polychlorinated biphenyls by arctic soil microorganisms. Appl Environ Microbiol 63:3378–3384
Morita RY (1975) Psychrophilic Bacteria. Bacteriol Rev 39:144–167
Muyzer G, De Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Muyzer G, Hottenträger S, Teske A, Wawer C (1996) Denaturing gradient gel electrophoresis of PCR-amplified 16 S rDNA—a new molecular approach to analyze the genetic diversity of mixed microbial communities. Mol Microbial Ecol Man 3.4.4(1):1–23
Pelz O, Tesar M, Wittich RM, Moore ERB, Timmis KN, Abraham WR (1999) Towards elucidation of microbial community metabolic pathways: unrevealing the network of carbon sharing in a pollutant-degrading bacterial consortium by immunocapture and isotopic ratio mass spectrometry. Environ Microbiol 1:167–174
Peters KE, Moldowan JM (1993) The biomarker guide. Interpreting molecular fossils in petroleum and ancient sediments. Prentice Hall Inc., Englewood cliffs
Polz MF, Cavanaugh CM (1998) Bias in template-to-product ratios in multitemplate PCR. Appl Environ Microbiol 64:33724–33730
Raghukumar C, Vipparty V, David JJ, Chandramohan D (2001) Degradation of crude oil by marine cyanobacteria. Appl Microbiol Biotechnol 57:433–436
Rosenberg E, Legmann R, Kushmaro A, Taube R, Adler E, Ron EZ (1992) Petroleum bioremediation—a multiphase problem. Biodegradation 3:337–350
Sahm K, Knoblauch C, Amann R (1999) Phylogenetic affiliation and quantification of psychrophilic sulfate-reducing isolates in marine arctic sediments. Appl Environ Microbiol 65:3976–3981
Syutsubo K, Kishira H, Harayama S (2001) Development of specific oligonucleotide probes for the identification and in situ detection of hydrocarbon degrading Alcanivorax strains. Environ Microbiol 3:371–379
Thomas JM, Yordy JR, Amador JA, Alexander M (1986) Rates of dissolution and biodegradation of water-insoluble organic compounds. Appl Environ Microbiol 52:290–296
Von Witzingerode F, Göbel UB, Stackebrandt E (1997) Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev 21:213–229
Westlake DWS, Jobson A, Philippe R, Cook FD (1974) Biodegradability and crude oil composition. Can J Microbiol 20:915–928
Whyte LG, Hwari J, Zhou E, Bourbonnière L, Inniss WE, Greer CW (1998) Biodegradation of variable-chain-length alkanes at low temperatures by a psychrotrophic Rhodoccoccus sp. Appl Environ Microbiol 64:2578–2584
Whyte LG, Slagmann SJ, Pietrantonio F, Bourbonniére L, Koval SF, Lawrence JR, Inniss WE, Greer CW (1999) Physiological adaptations involved in alkane assimilation at a low temperature by Rhodococcus sp. strain Q15. Appl Environ Microbiol 65:2961–2968
Wick LY, Springdael D, Harms H (2001) Bacterial strategies to improve the bioavailability of hydrophobic organic pollutants. In: Stegmann R, Brunner G, Calmano W, Matz G (eds) Treatment of contaminated soil. Springer, Berlin Heidelberg New York, pp 203–217
Wick LY, de Munain AR, Springael D, Harms H (2002a) Responses of Mycobacterium sp. LB501T to the low bioavailability of solid anthracene. Appl Microbiol Biotechnol 58:378–385
Wick LY, Wattiau P, Harms H (2002b) Influence of the growth substrate on the mycolic acid profiles of mycobacteria. Environ Microbiol 4:612–616
Yakimov MM, Guiliano L, Crisafi E, Chemikova TN, Timmis KN, Golyshin PN (2002) Microbial community of a saline mud volcano at San Biagio-Belpasso, Mt. Etna (Italy). Environ Microbiol 4:249–256
Ziemke F, Höfle MG, Lalucat J, Roselló-Mora R (1998) Reclassification of Shewanella putrefaciens Owen‘s genomic group II as Shewanella baltica sp. nov. Int J Syst Bacteriol 48:769–774
Acknowledgements
Thanks are due to Hauke Trinks from Hamburg University of Technology for supplying the samples from Spitzbergen.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by K. Horikoshi
Rights and permissions
About this article
Cite this article
Deppe, U., Richnow, HH., Michaelis, W. et al. Degradation of crude oil by an arctic microbial consortium. Extremophiles 9, 461–470 (2005). https://doi.org/10.1007/s00792-005-0463-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-005-0463-2