
Abstract
Autism spectrum disorder, severe behaviour problems and duplication of the Xq12 to Xq13 region have recently been described in three male relatives. To describe the psychiatric comorbidity and dysmorphic features, including craniosynostosis, of two male siblings with autism and duplication of the Xq13 to Xq21 region, and attempt to narrow down the number of duplicated genes proposed to be leading to global developmental delay and autism. We performed DNA sequencing of certain exons of the TWIST1 gene, the FGFR2 gene and the FGFR3 gene. We also performed microarray analysis of the DNA. In addition to autism, the two male siblings exhibited severe learning disability, self-injurious behaviour, temper tantrums and hyperactivity, and had no communicative language. Chromosomal analyses were normal. Neither of the two siblings showed mutations of the sequenced exons known to produce craniosynostosis. The microarray analysis detected an extra copy of a region on the long arm of chromosome X, chromosome band Xq13.1–q21.1. Comparison of our two cases with previously described patients allowed us to identify three genes predisposing for autism in the duplicated chromosomal region. Sagittal craniosynostosis is also a new finding linked to the duplication.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Xq duplications in the Xq12 to Xq21 region (dup (X) (q12q21)) were first described in the 1980s in children [1–3] presenting with severe intellectual developmental disorder (SIDD), short stature, facial anomalies and, in many cases, hypoplastic genitals, and have been reported as both maternally inherited [1–8] and as de novo duplications [9–12]. Lugtenberg and co-workers [12] have summarised the features of the eleven published cases of Xq duplications with clinical findings. They found that six of the eleven individuals had been examined in terms of social interaction skills and five of these were classified as socially impaired [12]. Thode et al. [3] described two male siblings and their male maternal cousin with Xq duplication. The two brothers exhibited impaired social interaction and one of them was described as “a depressed, withdrawn child” at the age of 9 years, while the other, at age 5, was “anxious”, “prone to tears and not easily coaxed into co-operating”. The male cousin was considered to function socially at the time of the examination at 5 years of age. More recently, Kaya et al. [8] reported Xq duplication with developmental delay and autistic features in two brothers and their maternal male relative. By expression analysis of duplicated Xq genes, these researchers suggested possible autism-predisposing or autism-mediator genes including the NLGN3, AR, EFNB1, OPHN1, TAF1, GJB1 and MED12.
Materials and methods
Clinical case report
The current case report presents two brothers who both meet the criteria for Xq duplication and autistic disorder [13]. The family consists of two parents, an older affected brother, a healthy sister, and a younger affected brother. The father has short stature, 160 cm (−3.1 SD), and his seven siblings are all short. The mother has no siblings. She is 162 cm tall (−0.9 SD). She has hypothyreosis, which is treated with Levaxin medication. The mother has a bilateral hearing impairment from early age affecting the lower frequencies. She wears hearing aids bilaterally.
The data on the two male siblings are drawn from personal contacts and medical records (child neuropsychiatry, plastic surgery, clinical genetics, paediatric rehabilitation, and obstetrics). Both siblings were individually psychiatrically and physically assessed by child neuropsychiatrists, neuropsychologists and clinical geneticists.
Patient 1: older brother born in 1985
Patient 1 was born at 38 weeks after a pregnancy complicated by decreased foetal movements. His birth weight was 2,540 g (−2 SD), birth length 45.5 cm (−2 SD) and his Apgar scores were 6, 6 and 9 at 1, 5 and 10 min, respectively. During the first 6 months of life he required hospitalisation with nasogastric tube feeding due to feeding problems and poor weight gain.
Sagittal craniosynostosis was diagnosed at 2 months and he had reconstructive surgery at 6.5 months with secondary surgery at 3.5 years. After the initial surgery he developed seizures, which persisted. He also had a right femoral and iliac artery thrombosis that later produced a leg length discrepancy. He had persistent short stature and at age 21, his height was reported to be 157 cm (−3.5 SD). He was diagnosed with hypopituitarism.
The patient was hypotonic and inactive as an infant. Developmental delay was noted during the first few months of life. He walked at age 3 and never developed any communicative language. He has been hyperactive from 2 years of age. Stereotypies, narrow interests (watching the washing machine going round and round for hours, always insisting on carrying a ball in his hand), and difficulty of interacting with peers were noted by age 5.5 years. A neuropsychiatric evaluation at age 6.5 years showed that the boy met the criteria for autistic disorder [14] and SIDD. The examination took place at a specialised child neuropsychiatry clinic evaluating tertiary referrals of autism from all over Sweden. On the autism behaviour checklist (ABC) [15] patient 1 had a total score of 85 (>67 is considered to indicate autism with “high probability”). He exhibited poor social interaction, had no peers, and made contact by slapping other children. He avoided eye contact. The parents reported routines and rituals, and if these were interrupted he would throw a temper tantrum. He did not understand the concept of danger. At age 13 he developed severe self-mutilative behaviour, including biting himself and hitting hard against tables. As an adult he has moved to a group residence for individuals with SIDD.
Physical examination at age 11 was notable for small ears with prominent crus and underdeveloped tragus, an elliptical face characterised by bilateral ptosis, epicanthus inversus, highly marked eyebrows, wide medial parts of the eyebrows situated far above the eyes, blepharophimosis, floating forehead, broad nasal base and tip, smooth filtrum, full upper and lower lips and small, hyperextensible joints, small pits and skin dimples situated around the joints, thin hands and feet and feet with short halluces (Figs. 1, 2).

Photograph of patient 1, AP view

Photograph of patient 1, lateral view
CT scan of the brain at 11 months showed a small frontal parenchymal defect. A reassessment at age 6.5 years showed left frontal pathology, indicating status post infarction or haemorrhagia interpreted as secondary to the asphyxia after the operation in early childhood. A chromosomal analysis at age 11 was normal. Neurometabolic screening (amino acids in blood and urine, organic acids, total sialic acid, oligosaccharides and mucopolysaccharides in urine) at 21 years showed normal results.
Patient 2: younger brother born in 1995
Patient 2 was born at 37 gestational weeks after a pregnancy complicated by decreased foetal movements. His birth weight was 2,670 g (−1 SD) and birth length was 45 cm (−1.5 SD). He had feeding problems and poor weight gain. Sagittal craniosynostosis was observed neonatally and he had reconstructive surgery at 5 months.
Developmental delay was observed in the first few months after birth. From the age of 14 months, he started showing interest in spinning toys and watching the washing machine going round and round. He walked at 2 years of age and never developed a communicative language. At age 3.5 years he developed severe sleeping problems, which persisted. Neuropsychiatric evaluation at the same clinic where patient 1 was examined showed that patient 2, at 6 years, fulfilled the criteria for autistic disorder [13]. He had a total score of 64 on the ABC, corresponding to “borderline autism”. The boy had poor social interaction, and exhibited poor eye contact, stereotypies, severe hyperactivity, self-mutilation (biting his index and middle finger), fascination with spinning wheels, temper tantrums, and no concept of danger. Patient 2 also met the criteria for SIDD. Nocturnal diuresis has persisted throughout childhood. Patient 2 reached puberty at 12 years of age.
Physical examination at age four showed brachycephalic skull, elliptical face, highly marked eyebrows, wide medial parts of the eyebrows situated far above the eyes, ptosis, broad nasal base and tip, small ears with prominent crus, micrognathia, full upper and lower lips and tapering fingers. Like his older brother, patient 2 had small pits and dimples around the elbows, knees and shoulders (Figs. 3, 4).

Photograph of patient 2, AP view

Photograph of patient 2, lateral view
CT scan of the brain was normal. He received treatment with growth hormone from 11 months until 9 years of age, due to persistent short stature. His height was reported to be 150 cm (−4.2 SD) at age 16. Low testosterone levels have been observed. Skeletal survey showed delayed bone age. Neurometabolic screening showed normal results. Chromosomal analysis was normal, except for a pericentric inversion on chromosome 9 at p11; q13, also seen in the older brother and the father. This finding is considered to be a normal variant.
Results
To rule out mutations causing the craniosynostosis syndrome associated with Saethre-Chotzen, Crouzon and Muenke syndrome DNA sequencing of exon 1 (the only coding exon of the gene) in the TWIST1 gene, exon 8, 10 and 11 in the FGFR2 gene and exon 7 and 10 in the FGFR3 gene was performed. This procedure was based on a clinical flowchart regarding investigations of craniosynostosis and co-existing SIDD. From a clinical perspective it was considered relevant to rule out those exons that represent the vast majority of syndromes combining craniosynostosis and intellectual developmental disorder. There were no mutations in any of the sequenced exons.
A microarray analysis was performed on DNA from the mother and three siblings, the two brothers and one sister, using the GeneRChipHuman Mapping 250 K Nsp I Array from Affymetrix. The investigation comprises analysis of approximately 250,000 single nucleotide polymorphism markers (SNPs) distributed over chromosome 1–22 and X. The analysis detects sub-microscopic copy number variations in terms of loss or gain of genetic material with a practical resolution of approximately 100–200 kb. An extra copy of a region on the long arm of chromosome X, chromosome band Xq13.1–q21.1 was detected in the two brothers and the mother, but not in the sister. The region comprises approximately 10 Mbp and covers the base pair positions 70117418-79993681, encompassing 23 known genes. In the two brothers and the mother, the duplication was verified by multiplex ligase-dependent amplification (MLPA) analysis of the ATRX-gene (35 probes included in probemix P013, MRC-holland). No analyses regarding X chromosome inactivation status were performed since the data are only warranted in females.
Discussion
These two male siblings with dup (X) (q13.1–q21.1) illustrate a spectrum of physical and psychiatric problems, some of which have not previously been described in association with duplications of genes in the region of Xq12 to Xq21 [1–12] (Table 1).
With regard to psychiatric “comorbidity”, the two male siblings exhibited, in addition to autistic disorder, SIDD, temper tantrums, no communicative language, hyperactivity and self-mutilation. Some previous publications [12] have found individuals with dup (X) (q12q21) to be socially withdrawn, a feature that can be interpreted as one of the core symptoms of autistic disorder. Poor or no language has previously been reported and this impairment can be a core symptom of autism. No other specific autistic features such as impairment of non-verbal communication or restricted repetitive behaviour have been reported. According to Table 1 SIDD is usually present in individuals with duplications of genes in the region (X) (q12q21). Autism occurs in 20–21 % [16, 17], of all individuals with SIDD, but can probably be overlooked in many cases, which can explain the lack of previous reports of autism in combination with SIDD regarding duplications in the present region.
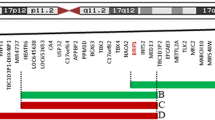
Recently, a novel X-linked disorder involving Xq12–q13.3 with developmental delay and autistic features was reported by Kaya and co-workers [8], who described three affected males, two siblings and their maternal grandmother’s cousin’s son. In addition to developmental delay in all three individuals, two of the males also had autistic traits. The break points of the duplication in the three male relatives (Xq12–q13.3) were slightly different from those observed in our cases (Xq13.1–q21.1), and significantly different from those of the cases described by Lugtenberg et al. [12], (Xq13.2–q21.1), which means that the duplicated region only partially overlaps in the three reports (see Fig. 5). The common overlapping region for all three reports is Xq13.2–Xq13.3, while the common overlapping region for the two reports describing autistic features is more centromeric: Xq13.1–Xq13.3. Hence, the cases described with autistic traits share common duplicated genes which are not duplicated in cases without autistic behaviour. By expression analysis and qRT-PCR of genes in the duplicated region, Kaya et al. found an increased gene dosage of several autism candidate genes in this chromosomal region. Three of the seven identified genes, MED12, NLGN3 and SLC16A2, are shared between our two patients and the three patients described by Kaya et al. [8]. One of these, the neuroligin 3 gene (NLGN3), shown by us to be associated with autism [18] and located in the region Xq12–q13.3, was suggested by Kaya’s group to be the cause of the autistic features in their case report. Neuroligin 3 is one of five neuroligins involved in the adhesion of glutamatergic synapses. Mutations of NLGN3 and the gene coding for neuroligin 4 (NLGN4) have both been found in individuals with autism [18] as well as in individuals with intellectual developmental disorder [19, 20]. NLGN3 is one of the three duplicated autism-predisposing genes in our two patients. Our finding of the duplicated NLG gene and earlier findings of duplication of the same gene indicate a gene dosage effect, i.e. the more gene product the more activity of the pathway where the gene is included. However, the relevance to autism of the two other genes, MED12 and SLC16A2, is less convincing, as both positive and negative associations with autism have been found for the MED12 gene [21, 22]. The SLC16A2 gene association with autism is even less convincing, as it is reported as an “autism-related disorder gene” [23]. However, both genes are implicated in signal transduction of the thyroid hormone and are known to produce neurological aberrations.

Ideogram of X chromosome with the duplicated Xq-region in the three relevant reports (Lugtenberg et al. [12], Kaya et al. [8], the present study). Expanded view showing duplicated genes in the common overlapping region. The three genes implicated in autism are marked with an asterisk
Sagittal craniosynostosis has, to our knowledge, not previously been associated with dup (X) (q12q21). The Saethre-Chotzen, Crouzon and Muenke syndromes are craniosynostoses linked to mutations of the TWIST1 (only exon), FGFR1, FGFR2, or FGFR3 located on chromosomes 4, 7 and 10. Case reports of autism and other kinds of craniosynostoses have been published [24, 25], but none of the syndromes were linked to the X chromosome.
In conclusion, this case report has shown that dup (X) (q12q21) can present with autistic disorder, severe behaviour disturbances and craniosynostosis, in addition to the previously reported short stature, facial anomalies, genital hypoplasia and SIDD. The NLGN3 gene location in the (X) (q12q21) area strengthens the linkage between mutations of this gene and the autistic disorder seen in the two affected brothers in the present case report.
References
Steinbach P, Horstmann W, Scholz W (1980) Tandem duplication dup(X)(q13q22) in a male proband inherited from the mother showing mosaicism of X-inactivation. Hum Genet 54:309–313
Vejerslev LO, Rix M, Jespersen B (1985) Inherited tandem duplication dup(X) (q131–q212) in a male proband. Clin Genet 27:276–281
Thode A, Partington MW, Yip MY, Chapman C, Richardson VF et al (1988) A new syndrome with mental retardation, short stature and an Xq duplication. Am J Med Genet 30:239–250
Yokoyama Y, Narahara K, Tsuji K, Moriwake T, Kanzaki S et al (1992) Growth hormone deficiency and empty sella syndrome in a boy with dup(X) (q13.3–q21.2). Am J Med Genet 42:660–664
Apacik C, Cohen M, Jakobeit M, Schmucker B, Schuffenhauer S et al (1996) Two brothers with multiple congenital anomalies and mental retardation due to disomy (X)(q12–>q13.3) inherited from the mother. Clin Genet 50:63–73
Shapira M, Dar H, Bar-El H, Bar-Nitzan N, Even L et al (1997) Inherited inverted duplication of X chromosome in a male: report of a patient and review of the literature. Am J Med Genet 72:409–414
Hou JW (2004) Inherited tandem duplication of the X chromosome: dup(X)(q13.2–q21.2) in a family. Chang Gung Med J 27:685–690
Kaya N, Colak D, Albakheet A, Al-Owain M, Abu-Dheim N et al (2012) A novel X-linked disorder with developmental delay and autistic features. Ann Neurol 71:498–508
Cremers FP, Pfeiffer RA, van de Pol TJ, Hofker MH, Kruse TA et al (1987) An interstitial duplication of the X chromosome in a male allows physical fine mapping of probes from the Xq13–q22 region. Hum Genet 77:23–27
Muscatelli F, Verna JM, Philip N, Moncla A, Mattei MG et al (1992) Physical mapping of an Xq-proximal interstitial duplication in a male. Hum Genet 88:691–694
Cheng SF, Rauen KA, Pinkel D, Albertson DG, Cotter PD (2005) Xq chromosome duplication in males: clinical, cytogenetic and array CGH characterization of a new case and review. Am J Med Genet A 135:308–313
Lugtenberg D, de Brouwer AP, Oudakker AR, Pfundt R, Hamel BC et al (2009) Xq13.2q21.1 duplication encompassing the ATRX gene in a man with mental retardation, minor facial and genital anomalies, short stature and broad thorax. Am J Med Genet A 149A:760–766
APA (1994) Diagnostic and statistical manual of mental disorders, 4th edition (DSM-IV). American Psychiatric Press, Washington
APA (1987) Diagnostic and statistical manual of mental disorders, 3rd edition revised (DSM-III-R). American Psychiatric Press, Washington
Krug DA, Aric J, Almond P (1980) Behavior checklist for identifying severly handicapped individuals with high levels of autistic behavior. J Child Psychol Psychiatry 21:221–229
Nordin V, Gillberg C (1996) Autism spectrum disorders in children with physical and mental disability or both. I. Clinical and epidemiological aspects. Dev Med Child Neurol 38:297–313
Saemundsen E, Juliusson H, Hjaltested S, Gunnarsdottir T, Haldisdottir T, Hreidarsson S et al (2010) Prevalence of autism in an urban population of adults with severe intellectual disabilities—a preliminary study. J Intellect Disabil Res 54:727–735
Jamain S, Quach H, Betancur C, Rastam M, Colineaux C et al (2003) Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 34:27–29
Raymond FL (2006) X linked mental retardation: a clinical guide. J Med Genet 43:193–200
Bourgeron T (2009) A synaptic trek to autism. Curr Opin Neurobiol 19:231–234
Michaelis RC, Copeland-Yates SA, Sossey-Alaoui K, Skinner C, Friez MJ et al (2000) The HOPA gene dodecamer duplication is not a significant etiological factor in autism. J Autism Dev Disord 30:355–358
Beyer KS, Klauck SM, Benner A, Poustka F, Poustka A (2002) Association studies of the HOPA dodecamer duplication variant in different subtypes of autism. Am J Med Genet 114:110–115
Visser WE, Swagemakers SM, Ozgur Z, Schot R, Verheijen FW et al (2010) Transcriptional profiling of fibroblasts from patients with mutations in MCT8 and comparative analysis with the human brain transcriptome. Hum Mol Genet 19:4189–4200
Megarbane A, Hersh JH, Chouery E, Fabre M (2002) Craniosynostosis, telecanthus, scalp hair abnormalities, and sensorineural deafness in two sibs. Am J Med Genet 109:323–327
Morey-Canellas J, Sivagamasundari U, Barton H (2003) A case of autism in a child with Apert’s syndrome. Eur Child Adolesc Psychiatry 12:100–102
Acknowledgments
The authors would like to thank the two boys, patient 1 and patient 2, and their family for their very helpful cooperation, without which this study would not have been possible.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wentz, E., Vujic, M., Kärrstedt, EL. et al. A case report of two male siblings with autism and duplication of Xq13–q21, a region including three genes predisposing for autism. Eur Child Adolesc Psychiatry 23, 329–336 (2014). https://doi.org/10.1007/s00787-013-0455-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00787-013-0455-1