
Abstract.
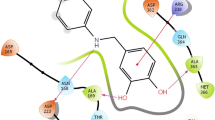
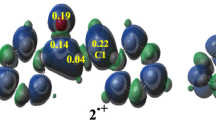
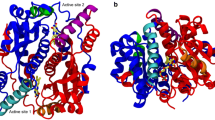
The viability of different mechanisms of catalysis and inhibition of the nickel-containing enzyme urease was explored using the available high-resolution structures of the enzyme isolated from Bacillus pasteurii in the native form and inhibited with several substrates. The structures and charge distribution of urea, its catalytic transition state, and three enzyme inhibitors were calculated using ab initio and density functional theory methods. The DOCK program suite was employed to determine families of structures of urease complexes characterized by docking energy scores indicative of their relative stability according to steric and electrostatic criteria. Adjustment of the parameters used by DOCK, in order to account for the presence of the metal ion in the active site, resulted in the calculation of best energy structures for the nickel-bound inhibitors β-mercaptoethanol, acetohydroxamic acid, and diamidophosphoric acid. These calculated structures are in good agreement with the experimentally determined structures, and provide hints on the reactivity and mobility of the inhibitors in the active site. The same docking protocol was applied to the substrate urea and its catalytic transition state, in order to shed light onto the possible catalytic steps occurring at the binuclear nickel active site. These calculations suggest that the most viable pathway for urea hydrolysis involve a nucleophilic attack by the bridging, and not the terminal, nickel-bound hydroxide onto a urea molecule, with active site residues playing important roles in orienting and activating the substrate, and stabilizing the catalytic transition state.
Article PDF
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Author information
Authors and Affiliations
Additional information
Electronic Publication
Rights and permissions
About this article
Cite this article
Musiani, F., Arnofi, E., Casadio, R. et al. Structure-based computational study of the catalytic and inhibition mechanisms of urease. J. Biol. Inorg. Chem. 6, 300–314 (2001). https://doi.org/10.1007/s007750000204
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s007750000204