
Abstract
In methylotrophic bacteria, which use one-carbon (C1) compounds as a carbon source, methanol is oxidized by pyrroloquinoline quinone (PQQ)-dependent methanol dehydrogenase (MDH) enzymes. Methylotrophic genomes generally encode two distinct MDHs, MxaF and XoxF. MxaF is a well-studied, calcium-dependent heterotetrameric enzyme whereas XoxF is a lanthanide-dependent homodimer. Recent studies suggest that XoxFs are likely the functional MDHs in many environments. In methanotrophs, methylotrophs that utilize methane, interactions between particulate methane monooxygenase (pMMO) and MxaF have been detected. To investigate the possibility of interactions between pMMO and XoxF, XoxF was isolated from the methanotroph Methylomicrobium buryatense 5GB1C (5G-XoxF). Purified 5G-XoxF exhibits a specific activity of 0.16 μmol DCPIP reduced min−1 mg−1. The 1.85 Å resolution crystal structure reveals a La(III) ion in the active site, in contrast to the calcium ion in MxaF. The overall fold is similar to other MDH structures, but 5G-XoxF is a monomer in solution. An interaction between 5G-XoxF and its cognate pMMO was detected by biolayer interferometry, with a KD value of 50 ± 17 μM. These results suggest an alternative model of MDH-pMMO association, in which a XoxF monomer may bind to pMMO, and underscore the potential importance of lanthanide-dependent MDHs in biological methane oxidation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Methylotrophs, bacteria that utilize one carbon (C1) compounds (devoid of carbon–carbon bonds) such as methane, methanol, and methylated amines as a carbon source, play a key role in the carbon cycle [1, 2]. Besides serving as the primary biological sink for methane and other methylated greenhouse gases, methylotrophs have been targeted as vehicles for bioremediation and production of fuels and chemicals [3, 4]. In these organisms, methanol is oxidized by methanol dehydrogenase (MDH) enzymes that use pyrroloquinoline quinone (PQQ) as a cofactor. The canonical MxaF-type MDHs have been studied extensively. MxaF-type MDHs comprise a large MxaF subunit (64 kDa) and a small MxaI subunit (8.5 kDa) [5]. The catalytic center, housed in MxaF, contains the PQQ cofactor and a calcium ion [6]. In the past two decades, a homolog of MxaF, XoxF, has also been implicated in methanol oxidation. MxaF and XoxF exhibit less than 50% amino acid sequence identity [5, 7], and xoxF genes are actually much more abundant than mxaF genes in methylotrophs, with some methylotroph genomes encoding only XoxF [7, 8].
Of particular relevance to bioinorganic chemistry is the recent discovery that XoxFs are dependent on the presence of lanthanide rather than calcium ions [9,10,11,12]. Lanthanides are a group of metals with atomic numbers 57–71 that are collectively referred to as rare earth elements (REEs) despite the fact that they are actually relatively abundant in the earth’s crust [7]. In initial studies, addition of La(III) and Ce(III) to methylotroph growth media was demonstrated to induce XoxF expression and promote growth on methanol [9,10,11]. In the case of methanotrophs, difficulties in culturing the Verrucomicrobial microbes isolated from Italian mudpots were solved by the addition of various REEs including La(III), Ce(III), Pr(III), and Nd(III) [12]. These lanthanides transcriptionally regulate the expression of MxaF and XoxF in a number of methylotrophs and methanotrophs [13,14,15,16]. For Methylomicrobium (Mm.) buryatense 5GB1C grown with 95 µM calcium in the medium, 1 µM of supplemental lanthanum was sufficient to abolish mxa transcription [15]. Since environmental lanthanum concentrations have been reported to be significantly higher than the amount shown to inhibit mxa transcription [17], XoxF is likely to be the functional MDH in many environments [7]. Consistent with the dependence on lanthanides, the only available XoxF structures, those of the Methylacidiphilum (Ma.) fumariolicum SolV enzyme (SolV-XoxF), reveal Ce(III), La(III), or Eu(III) ions in the active site [12, 18].
In methanotrophs, methylotrophs that utilize methane gas as their sole carbon source [19], the methanol substrate for MxaF and XoxF is produced by methane monooxygenase (MMO) enzymes. Both soluble and membrane-bound forms of MMO exist; the membrane-bound form, particulate MMO (pMMO), is predominant in nature [20]. Given the sequential action of pMMO and MDH in methanotroph metabolism, direct interactions between the two enzymes have been suggested and are supported by intracellular localization of MDH [21,22,23]. In addition, a putative pMMO-MDH supercomplex has been reported. Although purified samples of such a complex have not been obtained [24,25,26,27], specific protein–protein interactions between Methylococcus (Mc.) capsulatus (Bath) MxaF and pMMO have been detected [26]. Beyond providing a direct route for methanol from pMMO to MDH, a pMMO-MDH complex could also facilitate transfer of electrons from methanol oxidation back to pMMO via the electron acceptor of MDH, cytochrome cL [26]. The physiological reductant for pMMO remains unknown, but recent metabolic modeling for Mm. buryatense 5GB1C predicts that MDH may indeed mediate electron transfer to pMMO for methane oxidation [28], rather than the more widely accepted model involving NADH and a type 2 NADH:quinone oxidoreductase [29]. To gain further insight into potential pMMO-MDH interactions, particularly as pertains to the apparent prevalence of XoxF-type MDHs in the environment, we have isolated the lanthanum-containing XoxF from Mm. buryatense 5GB1C (5G-XoxF), determined its crystal structure, and investigated its interaction with Mm. buryatense 5GB1C pMMO (5G-pMMO).
Materials and methods
Growth of Mm. buryatense 5GB1C
Mm. buryatense 5GB1C cells were cultured as described previously [25]. Briefly, Mm. buryatense 5GB1C cells were grown in 12-L fermenter growths in 1× modified nitrate mineral salts (NMS2) medium (which contains 95 μM CaCl2·2H2O), 130 mM NaCl, 2.3 mM phosphate buffer, and 50 mM carbonate buffer, pH 9.5, supplemented with 30 μM LaCl3, 40 µM CuSO4 and 1× trace elements solution (500× is 1.0 g/L Na2·EDTA, 2.0 g/L FeSO4·7H2O, 0.8 g/L ZnSO4·7H2O, 0.03 g/L MnCl2·4H2O, 0.03 g/L H3BO3, 0.2 g/L CoCl2·6H2O, 0.02 g/L NiCl2·6H2O, 0.05 g/L Na2MoO4·2H2O). Cells cultivated for XoxF isolation were supplemented with 0.5% methanol. Approximately 10 g of frozen cell pellet were added to the fermenter to initiate growth at an optical density at 600 nm (OD600) of 0.1–0.2. All cells were cultured under an air-to-methane gas ratio of 3:1 at 30 °C and 300 rpm. Cells were harvested when the OD600 reached 10–11 and centrifuged for 30 min at 8000×g at 4 °C. Pelleted cells were flash frozen in liquid nitrogen and stored at − 80 °C.
Purification of XoxF from M. buryatense 5GB1C
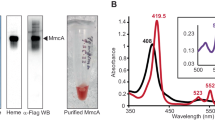
Mm. buryatense 5GB1C cell pellets (approximately 20 g) were resuspended in 200 mL of lysis buffer (25 mM PIPES, pH 7.3, 250 mM NaCl) and sonicated for 5 min (1 s on–off cycles at 40% amplitude) on ice. The lysed cells were centrifuged at 8000×g, followed by ultracentrifugation of the supernatant at 100,000×g to separate membranes from the soluble proteins. The supernatant was dialyzed overnight in 20 mM Tris, pH 8.0, 50 mM NaCl using a 10 kDa molecular weight cut off (MWCO) SnakeSkin dialysis tubing (ThermoFisher Scientific), and then loaded onto a Source 15Q anion exchange column (GE Healthcare). 5G-XoxF eluted at approximately 250 mM NaCl using a gradient of 50–400 mM NaCl in 20 mM Tris, pH 8.0. The fractions containing 5G-XoxF were collected and concentrated using an Amicon 30 kDa MWCO device (Millipore). After buffer exchanging into 20 mM Tris, pH 8.0, 250 mM NaCl, 5G-XoxF was loaded onto a Superdex 200 Increase 10/300 size exclusion analytical grade column (GE Healthcare) (Fig. S1). Fractions containing 5G-XoxF were concentrated to approximately 20 mg/mL, flash-frozen, and stored at − 80 °C. Sample purity was assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. S2), and protein concentration was measured with the Detergent-Compatible Lowry Assay (Bio-Rad) using bovine serum albumin (BSA) as a standard. The absorption spectrum of the purified enzyme at 40 μM was recorded in a UV-micro cuvette with a 1-cm path length (Chemglass) at room temperature on an Agilent 8453 UV–visible Spectroscopy instrument (Agilent Technologies) (Fig. S3). Inductively coupled plasma optical emission spectrometry (ICP-OES) and inductively coupled plasma mass spectrometry (ICP-MS) were performed at Northwestern University’s Quantitative Bio-element Imaging Center (QBIC), and lanthanum concentrations were quantified using 0–500 ppb lanthanum standards (Inorganic Ventures).
In-gel protein sequencing of 5G-XoxF
Protein identity was confirmed by in-gel protein sequencing mass spectrometry at Northwestern University’s Proteomics Core. Excised gel bands were washed in 100 mM ammonium bicarbonate (AmBic)/acetonitrile (ACN) and reduced with 10 mM dithiothreitol at 50 °C for 30 min. Cysteines were alkylated with 100 mM iodoacetamide in the dark for 30 min at room temperature. Gel bands were washed again in 100 mM AmBic/ACN prior to adding 600 ng trypsin for overnight incubation at 37 °C. The supernatant, which now contained peptides, was saved into a new tube. The remaining gel bands were then washed at room temperature for 10 min with gentle shaking in 50% ACN/5% formic acid (FA), and this solution was combined with the peptide solution. The wash step was repeated using 80% ACN/5% FA followed by 100% ACN. All supernatant was added to the peptide solution, which was then dried using a speed-vac. After lyophilization, peptides were reconstituted with 5% ACN/0.1% FA in water and injected onto a trap column (150 µm ID × 3 cm, in-house packed with ReproSil C18aq 3 µm) coupled with a Nanobore analytical column (75 µm ID × 10.5 cm, PicoChip column packed with ReproSil C18aq, 1.9 µm) (New Objectives, Inc., Woburn, MA). Samples were separated using a linear gradient of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in ACN) over 60 min using a Dionex UltiMate 3000 Rapid Separation nanoLC (ThermoFisher Scientific). MS data were obtained on a LTQ Velos Orbitrap (Thermo Fisher, San Jose, CA) mass spectrometer. The peptide sequences were compared to the UniProt Mm. buryatense 5G genome using Mascot 2.5.1 (Matrix Science, Boston, MA), and results were reported at 1% false discovery rate (FDR) in Scaffold 4.5 (Proteome Software, Portland, OR).
5G-XoxF methanol oxidation activity assay
Enzyme activity was measured by the 2,6-dichlorophenolindophenol (DCPIP) dye-linked dehydrogenase assay using phenazine methosulfate (PMS) (Sigma-Aldrich) as the mediator and methanol (Sigma-Aldrich) as the substrate as described previously [30]. Reactions were carried out at 30 °C in a 96-well clear, flat bottom, polystyrene Costar assay plate (Corning) containing 1.5 μM 5G-XoxF in a total reaction volume of 100 μL. A Biotek Cytation 5 imaging reader was used to measure the decrease in the absorbance at 600 nm which was monitored for 60 s. For determination of specific activity, change in A600 1 min after methanol addition was used to determine the concentration of reduced DCPIP. For Vmax, the maximum slope (A600/min) of reaction was used. The molar absorptivity of DCPIP at 600 nm is 1.91 × 104 M−1 cm−1.
Size exclusion chromatography with multi-angle light scattering (SEC-MALS)
The molecular mass of purified 5G-XoxF was determined using SEC-MALS. System components consist of an Agilent 1260 series high-performance liquid chromatography system (Agilent Technologies) for size exclusion chromatography equipped with a Wyatt Dawn Heleos II multi-angle static light scattering detector, a Wyatt QELS quasi-elastic (dynamic) light scattering detector, and a Wyatt T-rEx (refractometer with extended range) differential refractive index detector (all from Wyatt Technology, Santa Barbara, CA). 5G-XoxF at a concentration of 1, 5, and 10 mg/mL in 20 mM Tris, pH 8, 50 mM NaCl was injected onto a pre-equilibrated Superdex 200 10/300 GL column (GE Healthcare) with a flow rate of 0.5 mL/min at room temperature. ASTRA software (Wyatt Technology) was used to calculate the molecular mass, and BSA was used as a molar mass reference.
Purification of pMMO from Mm. buryatense 5GB1C
Cells grown for pMMO isolation were cultured and lysed as described above, but without 0.5% methanol supplementation. The pelleted membrane was homogenized in 25 mM PIPES, pH 7.3, 250 mM NaCl using a Dounce homogenizer, followed by centrifugation at 100,000×g for 30 min. This step was repeated twice for a total of three washes. The membranes were resuspended in 25 mM PIPES, 250 NaCl, pH 7.3 buffer to a final concentration of 10–20 mg/mL and flash frozen in liquid nitrogen for storage at − 80°C. The protein concentration was measured using the Detergent-Compatible Lowry Assay (Bio-Rad) with bovine serum albumin (BSA) as a standard.
Mm. buryatense 5GB1C membranes were solubilized in 1.2 mg n-dodecyl-β-d-maltopyranoside (DDM) (Anatrace) per mg of protein. The sample was centrifuged at 100,000×g for 30 min at 4 °C to pellet the membranes. The solubilized 5G-pMMO protein was then buffer exchanged with 25 mM PIPES, pH 7.3, 50 mM NaCl, 0.02% DDM using a 100,000 MWCO Amicon centrifugal concentrator (Milipore), and loaded onto a Source 15Q anion exchange column (GE Healthcare). 5G-pMMO eluted at approximately 320 mM NaCl using a gradient of 50–800 mM NaCl in 25 mM PIPES, pH 7.3, 0.02% DDM. Fractions containing 5G-pMMO were concentrated in an Amicon 100 kDa MWCO device (Millipore) to 10 mg/mL in 25 mM PIPES, pH 7.3, 250 mM NaCl, 0.02% DDM, and stored at − 80 °C. Sample purity was assessed using SDS-PAGE (Fig. S4). The 13C methane oxidation activity of the as-isolated membranes was 2.53 ± 0.34 nmol 13C methanol min−1 mg−1 protein (n = 3), performed as described previously [31]. The copper content of purified 5G-pMMO was 2.34 ± 0.18 copper equivalents per pMMO protomer (n = 3) using inductively coupled plasma optical emission spectroscopy (ICP-OES) at the Quantitative Bio-element Imaging Center (QBIC) at Northwestern University and 0–500 ppb copper standards (Inorganic Ventures).
Biolayer interferometry
Interactions between purified 5G-pMMO and purified 5G-XoxF were detected using a ForteBio biolayer interferometer (BLItz) in the Northwestern Keck Biophysics Facility. Amine reactive second-generation (AR2G) biosensors (ForteBio) were hydrated in 15 mM NaOAc, pH 5 for 30 min prior to each experimental run. This pH was chosen because it is slightly below the expected isoelectric point of 5G-XoxF (pI = 5.32), thus allowing electrostatic interactions to facilitate coupling between the sensor and the ligand. Activation of the sensor was carried out in four steps. First, the AR2G sensor was immersed in 15 mM NaOAc, pH 5 for 30 s to establish a baseline. Second, the biosensor was activated for 3 min in a mixture of 40 mg/mL EDC [1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide hydrochloride] and 10 mg/mL NHS [N-hydroxysuccinimide], both dissolved in water. For ligand loading, purified 5G-XoxF diluted to 0.5 mg/mL with 15 mM NaOAc, pH 5 was reacted with the biosensor for 3 min. Finally, any unbound sites were chemically blocked by reacting the sensor with 1 M ethanolamine, pH 8.5 for 3 min. After each step in the procedure, the sensor was immersed in baseline buffer for 1–2 min to wash away excess reagent from the previous step. The final prepared sensor was kept in baseline buffer before use.
Experimental runs were performed using 5G-pMMO diluted to concentrations of 5–25 μM in 15 mM NaOAc, pH 5 and 0.02% DDM. Each experimental run comprised three steps: (1) baseline, in which the 5G-XoxF-loaded sensor was immersed in pMMO buffer to establish a zero baseline for buffer interactions; (2) association, in which the sensor was immersed in purified 5G-pMMO for 2 min; and (3) dissociation, in which the sensor containing 5G-XoxF-pMMO complexes was transferred to baseline buffer for 3 min to dissociate. Each run was repeated with a new biosensor tip, and three total data sets were collected. One data set comprised five runs to test 5G-XoxF interactions with 0, 5, 10, 15, and 25 µM 5G-pMMO. The data were corrected against the 5G-pMMO buffer-only reference obtained at the start of each experimental set and fit with the global fitting function from ForteBio Data Analysis. A control experiment performed with BSA and 25 μM 5G-pMMO showed no interaction.
5G-XoxF structure determination
5G-XoxF crystals were obtained by the sitting drop vapor diffusion method by mixing 1 μL of 5 mg/mL XoxF with 1 μL of reservoir solution containing 0.2 M magnesium acetate, 0.1 M sodium cacodylate, pH 6.5, and 20% PEG 8000. Rod-shaped crystals formed after 1 week at room temperature. Data collection was performed at the Advanced Photon Source at Argonne National Laboratory. HKL2000 [32] was used to process and integrate all data sets. Phaser [33] was used to obtain molecular replacement solutions using the structure of XoxF from Ma. fumariolicum SolV (PDB accession code 4MAE) [12] as the search model (51% amino acid sequence identity). Starting from the initial model, COOT [34] was used to manually build the structure, followed by refinement using Phenix [35]. The model quality was assessed using MolProbity [36]. The final model for the 5G-XoxF structure includes residues 27–570 and 599–617, 1 La(III) ion, 1 PQQ molecule, 1 sodium ion, and 497 water molecules. The server PDBsum was used to identify hydrogen bonds (3.4 Å cutoff) at the dimer interfaces of 5G-XoxF and SolV-XoxF [37].
Results and discussion
Isolation and purification of XoxF from Mm. buryatense 5GB1C
In Mm. buryatense 5G, only one xoxF (METBUDRAFT_3845) is encoded in the xox operon, which also contains xoxF and xoxJ. Purification of 5G-XoxF has not been reported previously. In this study, 5G-XoxF was isolated from the soluble fraction, and supplementing the growth media with 0.5% methanol increased the growth rate by 40% and the yield of purified protein by tenfold. Similar effects of methanol supplementation were observed in previous XoxF studies [9, 11]. During purification, 5G-XoxF can be monitored by the absorbance at 345 nm that corresponds to its PQQ cofactor (Fig. S3). After two column chromatography steps (anion exchange and size exclusion), the yield of purified 5G-XoxF is typically ~ 50 mg/L of cell culture. The protein identity was confirmed by in-gel protein sequencing, and ICP-OES measurements indicated the presence of 0.7 ± 0.1 La(III) ions per monomer (n = 3). Purified 5G-XoxF exhibits methanol oxidation activity of 0.16 ± 0.05 μmol DCPIP reduced min−1 mg−1 (n = 3). This value is comparable to activity of MxaF-type MDH from Mc. capsulatus (Bath), but one order of magnitude less than values found for other characterized XoxFs (Table 1).
Solution oligomerization state of 5G-XoxF
The oligomeric state of purified 5G-XoxF was investigated via SEC-MALS at concentrations of 1, 5, and 10 mg/mL. The molecular weight of 5G-XoxF is 67.2 kDa, and for all samples, 5G-XoxF eluted as a single peak corresponding to 66 kDa (Fig. 1). Therefore, unlike other MDHs, 5G-XoxF exists predominantly a monomer. All other MDHs are homodimers and that from Mc. capsulatus (Bath) forms higher order oligomers [9, 11, 12, 26, 38]. One exception is XoxF from Candidatus Methylomirabilis oxyfera, which was purified as a XoxF/MxaI heterotetramer [38]. In addition, the XoxF from Methylobacterium extorquens AM1 was initially reported to be a monomer based on size exclusion chromatography [39], but was later purified from the same organism and reported to be a homodimer [11].

Solution oligomerization state of 5G-XoxF. Signals from the MALS refractive index detector are shown as a function of elution time (green for 1 mg/mL, blue for 5 mg/mL, and red for 10 mg/mL). The black horizontal lines indicate the calculated molecular masses of the eluting peaks
It is possible that the oligomerization state of XoxF is not as strictly conserved as that of MxaF, which is an important consideration for studying the structural aspects of its interaction with pMMO. In particular, a 16 Å resolution cryoelectron microscopy structure of a complex containing Mc. capsulatus (Bath) pMMO and MxaFI was interpreted to comprise an α3β3γ3 pMMO trimer interacting with an α3β3 trimer of MxaFI [27]. However, our later crystal structure of Mc. capsulatus (Bath) MxaFI revealed a dimer, and a stable complex between purified pMMO and MxaFI could not be isolated [26]. Instead, we hypothesized that multiple MDH dimers might transiently assemble into a “bilayer” with the small positively charged MxaI subunit facilitating interaction with the stacked intracytoplasmic membrane structures that house pMMO. However, this model is not applicable to XoxF since it lacks the second subunit, and the observed monomeric state of 5G-XoxF may suggest that in the cell, the monomeric form interacts with pMMO as originally suggested for the Mc. capsulatus (Bath) proteins [27].
Interaction between XoxF and pMMO from Mm. buryatense 5GB1C
The possibility of protein–protein interactions between the 5G-XoxF monomer and purified 5G-pMMO was investigated using biolayer interferometry. A concentration-dependent interaction was observed between the two proteins (Fig. 2), yielding a KD value of 50 ± 17 μM. By contrast, a KD value of 9.0 ± 7.7 μM was measured for the interaction between pMMO and MxaF from Mc. capsulatus (Bath) [26]. Not surprisingly, we were not able to detect a stable complex between the Mm. buryatense 5GB1C proteins by size exclusion chromatography, similar to the Mc. capsulatus (Bath) proteins [24, 26, 40], and consistent with the inability to enhance pMMO propylene epoxidation activity by combining purified proteins [24, 40]. As suggested previously, the association may be transient and/or facilitated by other components [26]. In addition, the weaker interaction could be characteristic of XoxF compared to MxaF or may be specific to the Mm. buryatense 5GB1C proteins. Analyses of multiple pMMO-MDH pairs is needed to further investigate the interaction between XoxF and pMMO. Regardless, these results indicate that the interaction with pMMO is not confined to MxaF-type MDHs, and does not require the presence of the small MxaI subunit.

Biolayer interferometry sensorgrams of the interaction between immobilized 5G-XoxF and 5G-pMMO. a Purified 5G-pMMO samples at concentrations of 5, 10, 15, and 25 µM were tested for binding to immobilized 5G-XoxF. The fitting curves are displayed in blue. b A control experiment testing the interaction between immobilized BSA and 25 µM 5G-pMMO was performed. No binding was observed. Each experimental run was repeated a total of three times with new sensors and protein samples
Structure of XoxF from Mm. buryatense 5GB1C
The structure of 5G-XoxF was determined to 1.85 Å resolution (Table 2). The asymmetric unit contains one molecule (Fig. 3a). The first residue in the structure is Asn 27, due to cleavage of the periplasmic targeting sequence, and the C-terminal residue is Asn 617, with no visible electron density for the residues between Ala 571 and Arg 598 (Fig. 3b). Like other MDHs, the monomer consists of eight sets of four-stranded antiparallel beta sheets, forming a beta propeller (Fig. 3a), which is surrounded by several short alpha helices. The N- and C-termini are found on the side of the protein opposite to the active site. 5G-XoxF exhibits 51% sequence identity to SolV-XoxF and 52% sequence identity to Mc. capsulatus (Bath) MxaF. Superimposing the main chain of 5G-XoxF with Mc. capsulatus (Bath) MxaF reveals minor differences in secondary structure with an rmsd of 0.69 Å for 574 Cα atoms. Substitutions mainly occur in loop regions on the protein surface, including a prominent loop in 5G-XoxF spanning residues Lys 469 to Gly 478. In addition, residues Lys 238 to Pro 244 form a beta sheet compared to a loop-helix structure in MxaF. Interestingly, these deviations seem to be specific to 5G-XoxF since they are also observed upon comparison to SolV-XoxF, which can be superposed on 5G-XoxF with a rmsd of 0.73 Å for 578 Cα atoms (Fig. 4).

Crystal structure of Mm. buryatense 5GB1C XoxF. a 5G-XoxF monomer. The eight-bladed β-sheet propeller forms a central cavity containing a PQQ ligand (blue) and a La(III) ion (yellow). A sodium ion from the solvent is modeled as a cyan sphere. b Disordered C-terminus of 5G-XoxF. Disordered residues 571-598 are represented with a dashed line (black). c 5G-XoxF active site. The 2Fo–Fc electron density map contoured at 1σ (gray) is shown for the La(III) ion, PQQ cofactor, and coordinating residues (pink). The anomalous difference Fourier map calculated using data collected at the La absorption edge (6.3 keV) is shown contoured at 10σ (green)

XoxF structure comparison. 5G-XoxF with SolV-XoxF (PDB accession code 4MAE) (both gray), with differing secondary structure elements shown in magenta for 5G-XoxF and dark blue for SolV-XoxF. a The disordered residues 571–598 of 5G-XoxF are modeled with α-helices in SolV-XoxF. b Residues Lys 469-Gly 478 form a prominent loop in 5G-XoxF, and residues Lys 238-Pro 244 in 5G-XoxF form a beta hairpin compared to a loop-helix structure in SolV-XoxF
Strong electron density observed adjacent to the PQQ cofactor, which is well defined, corresponds to a 35σ peak in the anomalous difference Fourier map generated with data collected at the La(III) absorption edge (6.3 keV). The density was well modeled with a La(III) ion at an occupancy of 0.8 (Fig. 3c), consistent with the metal analysis. The PQQ is sandwiched between Trp 267 and a disulfide bond formed between Cys 129 and Cys 130, residues that are conserved in other MDH structures (Fig. 3c) [6, 26, 41]. Hydrogen bonds in the active site stabilize the PQQ cofactor, similar to those in other MDH structures. Residue Asn 420 that interacts with the PQQ in 5G-XoxF is replaced by an aspartic acid in SolV-XoxF, but all other residues interacting with the metal and PQQ in the active site are conserved between the two XoxF structures. Similar to the Ce(III) ion in SolV-XoxF [12], the La(III) ion is 9-coordinate, ligated by the C-7 carboxylate, C-5 carbonyl, and N-6 quinoline nitrogen of the PQQ, as well as residues Glu 197 (bidentate), Asn 285, Asp 327 (monodentate), and Asp 329 (bidentate) (Table 3). In MxaF-type MDHs, the residue equivalent to Asp 329 is an alanine, and the Ca(II) ion is 6-coordinate [26]. As in SolV-XoxF, additional residue substitutions help accommodate the La(III) ion. Coordinating residue Asn 285 is positioned further away from the metal center due to the position of Thr 288, which replaces a proline found in MxaF. Similar alterations are observed near Glu 197 with Gly 196 replacing an alanine and Phe 198 replacing a leucine.
The active site appears more exposed than in other MDH structures due to the disordered residues near the C-terminus (Fig. 3b), which are stabilized by crystal contacts in the SolV-XoxF structure [12]. 5G-XoxF residues 575-589 are not conserved in SolV-XoxF and Mc. capsulatus (Bath) MxaF; there is a three amino acid (Asn 597, Ser 598, and Glu 599) insertion, which may contribute to the observed disorder. In addition, there is a negatively charged surface patch encapsulating the active site and neighboring the disordered region (Fig. 5a). These regions may interact with the positively charged surface of cytochrome cL for electron transfer [42], although cross-linking studies suggest interactions between lysine residues on MDH and carboxylate groups on cytochrome cL [43, 44]. Alternatively, these disordered residues could require other binding partners for stabilization. MxaJ is a periplasmic protein that is believed to serve a chaperone-like function in MDH assembly [45]. However, MxaJ has also been proposed to mediate interactions between MDH and its cytochrome cL electron acceptor [42, 46] or even between pMMO and MDH [47]. In the XoxF system, this role could be filled by the MxaJ homolog XoxJ, a protein of unknown function encoded in the xoxF operon.

Surface electrostatic potentials of 5G-XoxF and pMMO from Mm. alcaliphilum 20Z. Negatively charged surfaces are represented in red and positively charged surfaces in blue. a A 5G-XoxF crystallographic dimer, with one monomer represented as a surface. Black circles highlight the negative patch surrounding active site and disordered residues 571–598, and the positive patch at the XoxF dimer interface. b The negatively charged surface of the soluble domain of PmoB subunit from Mm. alcaliphilum 20Z pMMO (PDB accession code 6CXH) is circled in black
Although 5G-XoxF is a monomer in solution (Fig. 1), the dimer observed in other MDH structures is present in the crystal, mediated by crystal lattice contacts with a symmetry-related molecule (Fig. S6). This observation suggests that 5G-XoxF may be able to dimerize in solution, but perhaps at much higher concentrations than reported for other MxaFs and XoxFs (2.5–20 mg/mL) [12, 26]. Due to difficulty in obtaining high concentrations of purified protein from the native organism, samples at > 10 mg/mL were not analyzed by SEC-MALS. Similar to other MDH dimeric structures, the crystallographic dimer interface of 5G-XoxF forms a saddle shaped structure via β strands from residues 67–73 and 109–115 (Fig. S6). There are 14 hydrogen bonds at the interface, in comparison to 18 intermonomer hydrogen bonds in SolV-XoxF, and 30 intermonomer hydrogen bonds in MxaF from Mc. capsulatus (Bath) [26]. Notably, 5G-XoxF lacks two key hydrogen bonding pairs found in SolV-XoxF, Tyr 572 OH-Glu 570 OE2 and Tyr 572 OH-Ser 45 OG, which link β strands comprising residues 4–47 and 570–575 from each monomer. In 5G-XoxF, these three residues are substituted with threonine and positioned too far apart for polar contacts (6–9 Å). Furthermore, SolV-XoxF contains an additional C-terminal glutamic acid residue, Glu 577, which interacts with Arg 41. The lack of these contacts may account for the monomeric behavior of 5G-XoxF in solution. Another difference between 5G-XoxF and SolV-XoxF is the presence of a salt bridge between Glu 114 and Lys 605 in the 5G-XoxF structure; these residues are not conserved in SolV-XoxF.
Surface analysis of 5G-XoxF provides some insight into its interaction with pMMO. The PmoB subunit of Mm. alcaliphilum 20Z pMMO (20Z-PmoB) is 95% identical to 5G-pMMO PmoB [31]. MDHs are periplasmic [21,22,23] and in the case of the Mc. capsulatus (Bath) pMMO, PmoB interacts with pMMO [26]. The surface of 20Z-PmoB exhibits a prominent patch of negatively charged residues conserved in 5G-pMMO (Fig. 5b), which are complementary to and could potentially interact with the positively charged residues at the 5G-XoxF dimer interface (Fig. 5a). It is possible that these surfaces are involved in the protein–protein interactions observed by biolayer interferometry (Fig. 2).
Existence of multiple MDHs in Mm. buryatense 5GB1C
The Mm. buryatense 5G genome encodes both MxaF- and XoxF-type MDHs, a trend observed in many methylotrophs [8]. XoxF is found in a wider range of bacterial phyla compared to MxaF, and this phylogenetic diversity suggests more environmental importance for REEs than assumed previously [1]. While it has been shown that lanthanides downregulate MxaF expression in Mm. extorquens AM1, Methylosinus trichosporium OB3b, and Mm. buryatense 5G [14, 15, 48], the significance of having two MDH systems requiring different metals is unknown. The two MDH types may contribute to the modularity of C1 metabolism, which helps the organisms to withstand environmental fluctuations [8]. In addition, an active heterotetrameric complex consisting of XoxF with calcium and MxaI has been described, suggesting a functional relationship between the two systems [38]. The separation of XoxF into several phylogenetic groups (XoxF1–XoxF5) in NC10, Proteobacteria, Verrucomicrobia, and Methylophilaceae suggests that the role of lanthanides in regulation is complex [1, 5]. These considerations will be important for future work in studying lanthanide-dependent MDHs in C1 metabolism and as models for other yet-to-be discovered REE-dependent metalloenzymes.
Abbreviations
- ICP-MS:
-
Inductively coupled plasma mass spectrometry
- ICP-OES:
-
Inductively coupled plasma optical emission spectrometry
- Mc.:
-
Methylococcus
- MDH:
-
Methanol dehydrogenase
- Mm.:
-
Methylomicrobium
- MMO:
-
Methane monooxygenase
- pMMO:
-
Particulate methane monooxygenase
- PQQ:
-
Pyrroloquinoline quinone
- REE:
-
Rare earth element
- SEC-MALS:
-
Size exclusion chromatography with multi-angle light scattering
References
Chistoserdova L, Kalyuzhnaya MG (2018) Trends Microbiol. 26:703–714. https://doi.org/10.1016/j.tim.2018.01.011
Chistoserdova L, Lidstrom ME (2013) In: Rosenberg E, DeLong EF, Lory S, Stakebrandt E, Thompson F (eds) The prokaryotes. Springer, Berlin Heidelberg, pp 267–285
Strong PJ, Xie S, Clarke WP (2015) Environ Sci Technol 49:4001–4018
Pfeifenschneider J, Brautaset T, Wendisch VF (2017) Biofuels Bioprod Bioref 11:719–731
Keltjens JT, Pol A, Reimann J (2014) Op den Camp HJM. Appl Microbiol Biotechnol 98:6163–6183
Anthony C, Williams P (2003) Biochim Biophys Acta 1647:18–23
Skovran E, Martinez-Gomez NC (2015) Science 348:862–863
Chistoserdova L (2011) Environ Microbiol 13:2603–2622
Hibi Y, Asai K, Arafuka H, Hamajima M, Iwama T, Kawai K (2011) J Biosci Bioeng 111:547–549
Fitriyanto NA, Fushimi M, Matsunaga M, Pertiwiningrum A, Iwama T, Kawai K (2011) J Biosci Bioeng 111:613–617
Nakagawa T, Mitsui R, Tani A, Sasa K, Tashiro S, Iwama T, Hayakawa T, Kawai K (2012) PLoS One 7:e50480
Pol A, Barends TRM, Dietl A, Khadem AF, Eygensteyn J, Jetten MSM (2014) Op den Camp HJM. Environ Microbiol 16:255–264
Masuda S, Suzuki Y, Fujitani Y, Mitsui R, Nakagawa T, Shintani M, Tani A (2018) mSphere 3:e00462–17
Haque MFU, Kalidass B, Bandow N, Turpin EA, DiSpirito AA, Semrau JD (2015) Appl Environ Microbiol 81:7546–7552
Chu F, Lidstrom ME (2016) J Bacteriol 198:1317–1325
Vu HN, Subuyuj GA, Vijayakumar S, Good NM, Martinez-Gomez NC, Skovran E (2016) J Bacteriol 198:1250–1259
Bentlin FRS, Pozebon D (2010) J Braz Chem Soc 21:627–634
Jahn B, Pol A, Lumpe H, Barends TRM, Dietl A, Hogendoorn C, Op den Camp HJM, Daumann LJ (2018) ChemBioChem 19:1–8
Hanson RE, Hanson TE (1996) Microbiol Rev 60:439–471
Sirajuddin S, Rosenzweig AC (2015) Biochemistry 54:2283–2294
Wadzinski AM, Ribbons DW (1975) J Bacteriol 122:1364–1374
Fassel TA, Buchholz LA, Collins MLP, Remsen CC (1992) Appl Environ Microbiol 58:2302–2307
Brantner C, Remsen C, Owen H, Buchholz L, Collins M (2002) Arch Microbiol 178:59–64
Kitmitto A, Myronova N, Basu P, Dalton H (2005) Biochemistry 44:10954–10965
Puri AW, Owen S, Chu F, Chavkin T, Beck DAC, Kalyuzhnaya MG, Lidstrom ME (2015) Appl Environ Microbiol 81:1775–1781
Culpepper MA, Rosenzweig AC (2014) Biochemistry 53:6211–6219
Myronova N, Kitmitto A, Collins RF, Miyaji A, Dalton H (2006) Biochemistry 45:11905–11914
de la Torre A, Metivier A, Chu F, Laurens LML, Beck DAC, Pienkos PT, Lidstrom ME, Kalyuzhnaya MG (2015) Microb Cell Fact 14:1–15
Choi D-W, Kunz RC, Boyd ES, Semrau JD, Antholine WE, Han JI, Zahn JA, Boyd JM, de la Mora AM, DiSpirito AA (2003) J Bacteriol 185:5755–5764
Day DJ, Anthony C (1990) Methods Enzymol 188:210–216
Ro SY, Ross MO, Deng Y, Batelu S, Lawton TJ, Hurley JD, Stemmler TL, Hoffman BM, Rosenzweig AC (2018) J Biol Chem 293:10457–10465. https://doi.org/10.1074/jbc.RA118.003348
Otwinowski Z, Minor W (1997) Methods Enzymol 276:1–20
McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) J Appl Cryst 40:658–674
Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Acta Crystallogr Sect D Biol Crystallogr 66:486–501
Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) Acta Crystallogr Sect D Biol Crystallogr 66:213–221
Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2009) Acta Crystallogr Sect D Biol Crystallogr 66:12–21
Laskowski RA (2000) Nucleic Acids Res 29:221–222
Wu ML, Wessels HJCT, Pol A, Op den Camp HJM, Jetten MSM, van Niftrik L, Keltjens JT (2015) Appl Environ Microbiol 81:1442–1451
Schmidt S, Christen P, Kiefer P, Vorholt JA (2010) Microbiology 156:2575–2586
Basu P, Katterle B, Andersson KK, Dalton H (2003) Biochem J 369:417–427
Anthony C (2004) Arch Biochem Biophys 428:2–9
Nojiri M, Hira D, Yamaguchi K, Okajima T, Tanizawa K, Suzuki S (2006) Biochemistry 45:3481–3492
Chan HTC, Anthony C (1991) Biochem J 280:139–146
Cox JM, Day DJ, Anthony C (1992) Biochim Biophys Acta 1119:97–106
Van Spanning RJM, Wansell CW, De Boer T, Hazelaar MJ, Anazawa H, Harms N, Oltmann LF, Stouthamer AH (1991) J Bacteriol 173:6948–6961
Anthony C (1992) Biochim Biophys Acta 1099:1–15
Choi JM, Cao T-P, Kim SW, Lee KH, Lee SH (2017) Proteins Struct Funct Bioinf 85:1379–1386
Skovran E, Palmer AD, Rountree AM, Good NM, Lidstrom ME (2011) J Bacteriol 193:6032–6038
Page MD, Anthony C (1986) J Gen Microb 132:1553–1563
Goodwin MG, Anthony C (1996) Biochem J 318:673–679
Acknowledgements
This work was supported by NIH Grant GM118035 (A.C.R.) and a grant from the Undergraduate Research Grant Program which is administered by Northwestern University’s Office of Undergraduate Research. The authors thank Dr. Mary Lidstrom at Washington University for providing Mm. buryatense 5GB1C cultures and Theint Aung from the Northwestern Keck Biophysics Facility for assistance with BLItz instrumentation, data collection, and data interpretation. This work used the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). Data were collected at the LS-CAT beamlines 21-ID-D/F/G. Use of GM/CA has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). The GM/CA Eiger 16 M detector at beamline 23ID-B was funded by an NIH-Office of Research Infrastructure Programs, High-End Instrumentation Grant (1S10OD012289-01A1).
Author information
Authors and Affiliations
Corresponding author
Additional information
PDB accession code: 6DAM.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Deng, Y.W., Ro, S.Y. & Rosenzweig, A.C. Structure and function of the lanthanide-dependent methanol dehydrogenase XoxF from the methanotroph Methylomicrobium buryatense 5GB1C. J Biol Inorg Chem 23, 1037–1047 (2018). https://doi.org/10.1007/s00775-018-1604-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-018-1604-2