
Abstract
The aim of this work is to understand the potential health effects of metal nanoparticles by exposing human leukemic cell lines (jurkat, K562 and KG1A cells) to nanosize phosphonomethyl iminodiacetic acid coated cobalt oxide (PMIDA-CoO) NPs. The synthesized PMIDA-CoO NPs were characterized by XRD, dynamic light scattering, transmission electron microscopy and scanning electron microscopy. Our results showed that exposure of leukemic cell lines to PMIDA-CoO NPs caused reactive oxygen species (ROS) generation by increasing the concentration of free Co++ ions in cancer microenvironment. But at physiological pH, PMIDA-CoO liberates little amount of Co++ ions into media and exerts lower toxicity to normal cells up to a certain dose. PMIDA-CoO NPs caused DNA damage in leukemic cell lines, which was reflected by an increase in apoptosis of jurkat, KG-1A and K562 cells. PMIDA-CoO NPs induced apoptosis by increasing pro-inflammatory cytokines, primarily TNF-α. The in vivo study shows that PMIDA-CoO NPs were efficiently killed DLA cells. These findings have important implications for understanding the potential anticancer property induced by surface-modified cobalt oxide nanoparticles.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The interesting magnetic and electrical properties along with the good chemical and thermal stabilities of metal nanoparticles are being utilized in many applications including magnetic extraction, magnetic resonance imaging, cell labeling, drug delivery and hyperthermia [1–4]. Some nanomaterials may have genotoxic or carcinogenic effects because of the metals used to make them. In addition, some metal particles can lead to reactive oxygen species (ROS) generation that causes oxidative stress and DNA damage in the body [5, 6].
Cobalt has a physiological role as a co-factor of vitamin B12, but cannot be regarded only as an essential element. Among cobalt-based nanoparticles, cobalt oxide nanoparticles in particular are currently attracting enormous interest due to their unique size and shape-dependent properties and potential applications, such as pigments, catalysts, sensors, electrochemistry, magnetism, and energy storage. [7]. Cobalt-based magnetic fluids designed for possible use in medical applications have been used because of higher magnetic properties and their greater effects on proton relaxation [8].
Cobalt oxide nanoparticles induced oxidative stress, DNA damage, genotoxic effects and inflammatory responses [57–59]. Azaria et al. [56] in 2011 hypothesized that the toxic effects of cobalt NPs are mainly due to cobalt ion dissolution from the nanoparticles. This group also proved that the cobalt oxide nanoparticle-induced toxicity depends on concentration and exposure time. Beside these, they also proved that primary DCs were the least sensitive cells among the other used cell lines (A549, MDCK, NCIH441, Caco-2 and HepG2).
Magnetic cobalt nanoparticles are used in drug delivery in eye surgery to repair detached retinas [9, 10]. Cobalt NPs are restricted in use because of their toxicity. Recent developmental work has largely focused on new polymeric or inorganic coatings on magnetite/maghemite nanoparticles [11]. This modification involves the chemical modification of the magnetic nanoparticle surface with organophosphorus compounds which offer a promising alternative in the coupling of organic components to metal oxides [12]. The bonding of organophosphorus molecules of the inorganic phase results from the formation of strong M–O–P bonds through heterocondensation and coordination. Homocondensation with the formation of P–O–P bridges is unlikely and such bridges are not stable in the presence of water. Organophosphorus coupling agents (OPCA) react specifically with metal oxide surfaces and assist only monolayer formation. The resulting monolayers are highly stable under physiological conditions [13–15].
Therefore, this study has been focused on the development of a potent anticancer agent for in vivo and in vitro study. We observed that the PMIDA-coated cobalt oxide nanoparticles (PMIDA-CoO NPs) have the ability to kill the cancer cells by releasing Co++ ions into cancer microenvironments and that Co++ ion-induced oxidative stress linked to activation of TNF-α-p38 MAPK and caspase-8-mediated death signals in human leukemia cells.
Materials and methods
Chemicals and reagents
Cobalt chloride (CoCl2·3H2O), phosphonomethyl iminodiacetic acid (PMIDA), propidium iodide (PI), RNaseA, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT reagent), rhodamine-B (RhB), histopaque 1077, TNF and IL-10 ELISA Kit, caspase-3, -8 p38, and pAKT antibodies were procured from Sigma (St. Louis, MO, USA) and eBiosciences, respectively. RPMI 1640, fetal bovine serum (FBS), penicillin, streptomycin, sodium chloride (Nacl), sodium carbonate (Na2CO3), sucrose, Hanks balanced salt solution and ethylene diamine tetra acetate (EDTA) were purchased from Himedia, India. Tris–Hcl, Tris buffer, KH2PO4, K2HPO4, Hcl, formaldehyde, alcohol and other chemicals were procured from Merck Ltd., SRL Pvt. Ltd., Mumbai, India. Commercially available dimethyl sulfoxide (DMSO) was procured from Himedia, India and purified by vacuum distillation over KOH. All other chemicals were from Merck Ltd., SRL Pvt., Ltd., and were of the highest purity grade available.
Synthesis of PMIDA-CoO NPs
PMIDA cobalt oxide nanoparticles were synthesized by the thermal decomposition method according to our previous lab report [51]. Two grams of starting material (CoCl2·3H2O) was taken in a beaker and mixed with Na2CO3 as a molar ratio 1:1 and the solution was rotated 1 h at room temperature. The precipitations were collected by centrifugation and were calcinated at 300 °C in air in a porcelain crucible for 2 h to get cobalt oxide NPs. A solution of synthesized cobalt oxide NPs (3 mg/ml) and PMIDA (prepared by dissolving 27 mg PMIDA in 10 ml of distilled water) were mixed and stirring for 12 h. The pH of the medium was maintained at 10. After that particles were collected by centrifugation. Then the recovered particles were washed three times with milli-Q (Millipore).
Characterization of the PMIDA-CoO NPs
X-ray diffraction study
The phase formation and crystallographic state of PMIDA-CoO NPs were determined by XRD with an Expert Pro (Phillips) X-ray diffractometer using CoKα radiation (¥ = 0.178 nm). Samples were scanned from 20° to 80° of \(2\theta\) increments of 0.04° with 2-s counting time. The hydrodynamic size of the PMIDA-CoO NPs aggregates was measured in a Brookhaven 90 Plus particle size analyzer [16].
Dynamic light scattering
The size distribution and zeta potential of the PMIDA-CoO NPs were determined by dynamic light scattering (Zetasizer Nano ZS; Malvern Instruments Ltd, Malvern, Worcestershire, UK) [16].
Zeta potential
The zeta potential of the PMIDA-CoO NPs was measured using a Zetasizer Nano ZS (Malvern, Malvern Hills, UK). 1 mg/ml PMIDA-CoO NPs solution was prepared in Milli-Q water. Then the solution was filtered using whatman No 1 filter paper and the filtrate was used to experiment [16].
Transmission and scanning electron microscopy
The particle size and microstructure were studied by high-resolution transmission electron microscopy in a JEOL 3010 (HRTEM), Japan, operating at 200 kV according to the method of Mohapatra et al. [17] with some modifications. Briefly, PMIDA-CoO NPs were suspended in deionized water at a concentration of 1 mg/ml, then the sample was sonicate using a sonicator bath until the sample was from a homogeneous suspension. For size measurement, sonicated stock solution of all PMIDA-CoO NPs (0.5 mg/ml) was diluted 20 times. TEM and SEM were used to characterize the size and shape of the PMIDA-CoO NPs. A drop of aqueous PMIDA-CoO NPs suspension was placed onto the carbon-coated copper grid and this was dried in the air to get TEM and SEM image.
FT-IR spectra
The conjugation of PMIDA with CoO NP was investigated by FTIR with a model PERKIN-ELMER SPECTRUM RXIFT-IR SYSTEM [17].
Dissolution study
Two concentrations of CoO NPs and PMIDA-CoO were (100 and 50 µg/ml) suspended in medium (without FBS and antibiotics) and incubated for 1 week at 37 °C. After the incubation period, the Co++ ions were separated from suspension of bare CoO NPs and PMIDA-CoO NP using the dialysis membrane [60]. The supernatants were used for the estimation of free cobalt ions in the medium by atomic absorption spectroscopy (AAS) using different concentrations of CoCl2·6H2O as a standard. The concentration of Co++ ions released from bare CoO NPs at 100 and 50 µg/ml were referred as U1 and U2 and PMIDA-CoO NPs was referred as U3 and U4, respectively, using different controls (0.001, 0.0005, 0.00025, 0.00001, 0.000005, and 0.000001 M). All measurements were analyzed in triplicate.
In vitro cytotoxicity to normal cells
The lymphocytes were isolated from heparinized blood samples according to the method of Hudson and Hay [18]. Normal human lymphocytes were divided into six groups. Each group contained six Petri dishes (2 × 104/ml cells in each). The cells of each Petri dish of control and experimental groups were maintained in RPMI 1640 media, supplemented with 10 % FBS, 50 μg/ml gentamycin, 50 μg/ml penicillin and 50 μg/ml streptomycin at 37 °C in a 95 % air and 5 % CO2 incubator. PMIDA-CoO NPs were added to the cells at different concentrations (1, 5, 10 25 and 50 µg/ml), were incubated for 24 h at 37 °C in a humidified incubator (NBS) and maintained with 5 % CO2, The cell viability was estimated by 3-(4,5-dimethylthiazol)-2-diphenyltetrazolium bromide (MTT) according to the method of our previous laboratory report [19]. The 24-h time points were selected according to a previous research which showed a high morphological transformation, reflecting a higher carcinogenic potential after 72-h incubation with cobalt oxide nanoparticles [58]. All measurements were analyzed in triplicate.
In vitro toxicity of PMIDA-CoO NPs on leukemic cell lines
The jurkat, KG-1A and K562 cell lines were obtained from the National Centre for Cell Sciences (NCCS) Pune, India. Cell lines were cultured in RPMI 1640 medium supplemented with 10 % fetal calf serum, 100 units/ml penicillin and 100 µg/ml streptomycin, 4 mM l-glutamine under 5 % CO2 and 95 % humidified atmosphere at 37 °C. Jurkat, K562 and KG-1A cell lines (2 × 104/ml cells in each) were seeded into 96 wells of tissue culture plates having 180 µl of complete media and were incubated for 48 h. PMIDA-CoO NPs were added to the cells at different concentrations (1, 5, 10, 25 and 50 µg/ml), were incubated for 24 h at 37 °C in a humidified incubator (NBS) and maintained with 5 % CO2. The cell viability was estimated by 3-(4,5-dimethylthiazol)-2-diphenyltetrazolium bromide (MTT) according to the method of our previous laboratory report [19]. Doxorubicin was used as a positive control. All measurements were analyzed in triplicate.
Intracellular uptake
Nanoparticle uptake by jurkat cells, K562 cells, KG-1A cells and normal lymphocytes cells was studied by rhodamine-tagged PMIDA-CoO NPs and atomic absorption spectroscopy [20].
For fluorescence microscopy, the cells were treated with rhodamine-tagged PMIDA-CoO NPs in a humidified incubator maintained with 5 % CO2 at 37 °C. After 8-h incubation, fluorescence images were acquired with 488 nm laser for differential interference contrast microscopy and 543 nm lasers for RITC excitation on an Olympus research phase contrast with fluorescence microscope (Olympus Singapore Pvt. Ltd., Valley Point Office Tower, Singapore).
In another experiment, leukemic cells were (2 × 104 cells/well) cultured into 35 mm cell culture plates. They were incubated with PMIDA-CoO NPs in a humidified incubator maintained with 5 % CO2 and 37 °C. After 8 h, the cobalt (Co++) uptake in jurkat cells K562 and KG-1A after PMIDA-CoO NPs’ treatment was estimated by atomic absorption spectroscopy (AAS). AAS cells were harvested at 50 % confluence (2 × 104 cells) and then treated with PMIDA-CoO NPs at 50 µg/ml dose for 2, 4, 6 and 8 h. After the treatment schedule, cells were washed twice with equal volumes of PBS with or without 1 mM EDTA. The cells were resuspended in 6 M nitric acid and incubated at 95 °C for 24 h. Acid-digested samples were then assayed for cobalt content with a Shimadzu AA-7000 atomic absorption spectrophotometer. A standard curve of six standard samples (0.001, 0.0005, 0.00025, 0.00001, 0.000005, and 0.000001 M) was prepared using CoCl2·6H2O salt, dissolved in RPMI-1640 media. The final values of cellular cobalt content before and after PMIDA-CoO NPs treatment were estimated from the standard curve. Cobalt content of all buffers and culture medium was normalized to get the actual CO++ content of cells [20]. The assay was repeated 5 times.
Measurement of ROS
The production of intracellular ROS was measured using 2,7-dichlorofluorescin diacetate (DCFH2-DA) [21]. The DCFH2-DA passively enters the cell where it reacts with ROS to form the highly fluorescent compound dichlorofluorescein (DCF). In brief, 10 mM DCFH2-DA stock solution (in methanol) was diluted in culture medium without serum or other additive to yield a 100-μM working solution. At the end of exposure with PMIDA-CoO NPs and 100 µM H2O2 [55] (as a positive control), cells were washed twice with PBS. Then cells were incubated in 1.5-ml working solution of DCFH2-DA at 37 °C for 30 min. Cells were lysed in alkaline solution and centrifuged at 2,300×g. A 1-ml supernatant was transferred to a cuvette and fluorescence was measured using at 485 nm excitation and 520 nm emission using a fluorescence spectrophotometer (Hitachi F-1700). The values were expressed as percent of fluorescence intensity relative to control wells. Fluorescence micrographs were also taken using phase contrast microscopy.
Pretreatment with NAC
To determine the role of ROS in NP-induced cell death, jurkat cells were seeded in a 96-well plate at 0.2 ml per well at a concentration of 5 × 105 cells/ml. A stock solution of N-acetyl cysteine (NAC, Sigma-Aldrich) was made with sterile water and added to cells at 10 mM for 1 h [21]. After NAC pretreatment, cells were cultured with PMIDA-CoO NP for 24 h. Viability was determined by MTT method. 100 µM H2O2 was used as a positive control. All measurements were analyzed in triplicate.
DNA fragmentation detection
DNA damage in cancer cells with exposure to metal nanoparticles was examined by agarose gel electrophoresis. In brief, cells were seeded in culture flasks (3 × 105 cells per flask) and treated with 25 μg/ml of PMIDA–PMIDA-CoO NPs for 24 h. After treatment, the cells were resuspended at a concentration of 1 × 105 cells per ml of cold 50 mM PBS. DNA from each sample was extracted twice with phenol–chloroform–isoamyl alcohol (PCI, 25:24:1; v:v:v) and once with chloroform as described in the literature [22]. After isolation, the DNA pellet was dissolved in TE buffer (Tris–HCL 10 mmol/L, pH 7.4, ethylenediaminetetraacetic acid 1 mmol/L, pH-8.0). Before electrophoresis, the DNA concentration of samples was adjusted to approximately the equivalent concentration. Usually 2–3 mg of DNA sample was mixed with loading buffer and placed in the well of agarose gel. The gel agarose (1.5 %, w/v) in electrophoresis buffer (Tris-botalic-ethylene diaminetetraacetic acid buffer) was run at 100 V until the purple tracer marker migrated to ~2 cm before the end of the gel. The gels stained with ethidium bromide (Sigma) at a concentration of 2 mg/L were photographed under UV light using BioRad Gel Doc. All measurements were analyzed in triplicate.
Cytokine analysis
To investigate the effect of PMIDA-CoO NPs on cytokine production, an ELISA was used for determination of TNF-α, IL-10 production. The cancer cells were cultured at 1 × 105 cells/ml and treated with varying concentrations of PMIDA-CoO for 24 h. After NP treatment, cell-free supernatants were harvested via successive 10-min centrifugations (2,000 rpm, 7,000 rpm and 13,000 rpm) and stored at −80 °C until analysis. ELISA was performed by the eBio Sci. Kit with all samples analyzed in triplicate. LPS-treated cells were used as positive control. All measurements were analyzed in triplicate.
Ethidium bromide and acridine orange stain
Apoptosis or necrosis was assessed using ethidium bromide (EtBr) in combination with acridine orange (AO) staining [49]. After 12 h of incubation, the cells were isolated, and a solution of PBS containing EtBr and AO (50 µg/ml; Vol/Vol) was then added. Living cells were stained with green (AO). Only cells that had membrane pores or cells that had ruptured allowed the diffusion of EtBr into the cell cytosol. Images were acquired using an inverted fluorescence microscope with original magnification of 400×. All measurements were analyzed in triplicate.
Apoptotic markers in vitro
After the treatment schedule, cells were lysed and centrifuged to collect the supernatant. The supernatants were used for the detection of pro-apoptotic and anti-apoptotic markers using ELISA [54]. The plates were coated with caspase-8 (50 µl/well) and caspase-3 (50 µl/well) capture antibodies (2 mg/ml) diluted in 0.05 M carbonate buffer pH 9.6. After overnight incubation at 4 °C, plates were washed three times with 0.15 M PBS, 0.05 % Tween-20 (PBST) and blocked with 50 µl/well PBS, 5 % FBS, 0.05 % Tween-20, 0.02 % sodium azide (PBSTN) for 1 h at room temperature. The plates were washed with PBST for three times and 100 µl of samples was added to each well and incubated at room temperature for 2.5 h. Plates were washed four times with PBST and incubated with 50 µl/well of biotinylated anti-caspase-8 and caspase-3 detection antibody for 2 h at room temperature. After three washes with PBST, 50 µl/well of HRP-avidin solution (e Biosciences) was added. After 30 min at room temperature, the plates were washed twice in PBST and then 100 µl/well substrate buffer (e Biosciences) was added and incubated in the dark at room temperature for 30 min. Optical densities were then measured at 450 nm using an ELISA reader (BioRad). All samples were analyzed in triplicate. All measurements were analyzed in triplicate.
Immunofluorescent staining
The leukemic cells were treated with 25 µg/ml of PMIDA-CoO NPs for 24 h. At the end of treatment(s), the cells were washed with PBS, pH 7.2, and were fixed in 4 % paraformaldehyde solution for 15 min at 4 °C. Next, the cells were washed three times with the blocking buffer (5 % bovine serum albumin in PBS), followed by permeabilization with 0.5 % Triton X-100 in PBS for 20 min at 25 °C. After three washes with the blocking buffer, the cells were incubated for 1 h at 25 °C with mouse antibodies specific for caspase-8, -3, p38. The antibodies were used at dilutions of 1:100, in blocking buffer. The cells were then rinsed three times with the blocking buffer and probed in the dark with rhodamine-conjugated compatible secondary antibodies for 1 h at 25 °C. Following three washes with the blocking buffer, the coverslips were mounted on glass slides using a Fluoromount G (Electron Microscopy Sciences, Fort Washington, PA, USA). The slides were viewed using a fluorescence microscope (Nikon Eclipse E-800) equipped with the suitable wavelength filters [48].
Expression of phosphorylated p38, cleaved caspase-3 and caspase-8 determination by flow cytometry
In normal untreated and treated cell cultures, we determined the abcam anti-caspase and anti-p38 kit by mentioning the manufactures’ protocol and the method describe elsewhere [61]. Cells were resuspended in PBS and stained according to protocol to detecting protein or activation of the phosphorylation state. All the cells were incubated with permeabilizing buffer containing 0.2 % TritonX-100 and washed with PBS (1X) for three times. Then the cells were incubated with primary antibody for 1 h followed by anti-human antibody labeled with FITC and Texas red. For each sample, at least 20,000 events were acquired in a FACS caliber and versa, and the data were quantified using CellQuest software.
Induction of Dalton’s lymphoma (DL) in mice
The in vivo studies were conducted on female Swiss albino mice aged 5–6 weeks, weighing 25 ± 5 g, and housed in polycarbonate cages (five mice per cage) at an ambient temperature of 25 ± 2 °C with a 12-h light and a 12-h dark cycle. The mice were fed with commercially obtained rodent chow and water ad libitum. The animals were allowed to acclimatize to the laboratory environment and were then randomly subjected to the experiment. Mice were maintained under standard laboratory conditions, as per the guidelines and approval from the institutional animal ethical committee, with free access to commercially available food pellets and water. As described earlier [23, 24], Dalton lymphoma (DL) was induced by intraperitoneal (IP) serial transplantations of 1 × 107 viable tumor cells (assayed by the trypan blue method) per mouse with 100 % success each time. Development of DL was confirmed by abnormal belly swelling and increased body weight, which were visible in 10–12 days of implantation. The untreated DL mice survived for 18 ± 2 days.
Experimental design
The mice were divided into four groups, with six animals in each group: Group 1, blank non-tumor mice (non-tumor, untreated); Group 2, tumor control mice (tumor induced, untreated); Group 3 to Group 6, tumor-induced mice treated with PMIDA-CoO NPs at a concentration of 100, 200, 500 and 1000 μg/kg BW in PBS via intraperitoneal (IP) injection for 15 days at 3-day interval after the development of tumor. To assess the effects of compounds on the general appearance of DL mice, the body weights of the mice were recorded at an interval of 3 days starting from the day of transplantation up to 15 days. The mortality was noted in each group and increases in the survival time of mice of both the treated groups were analyzed using the following calculation:
where T number of days the treated animals survived and C number of days the control animals survived [25].
Euthanasia of experimental animals
After completion of the experimental treatment, all the mice were deprived of food overnight and euthanized by cervical dislocation under ketamine–xylazine anesthesia. The ascetic fluid and blood samples were collected carefully for different types of estimations. All measurements were analyzed in triplicate.
Effect of nanoparticles on ascetic tumor
The MTT assay was used for the in vivo experiments. Tumor mice in Group 3–6 were treated with PMIDA-CoO NPs at different concentration for a period of 15 days, and their ability to reduce tumor volume and the number of cells was compared with Group 2 tumor control mice. All measurements were analyzed in triplicate.
Assay for cell death
Ascetic fluid plays a crucial role in DLA and is a collection of pleomorphic cells with hyperchromatic nuclei that are clumps of malignant cells. The viability of tumor cells in ascetic fluid can lead to further aggravation of disease, and hence the morphology and viability of cells in ascetic fluid of the controls and tumor-treated mice were observed by cytological analysis and MTT assay. The ascetic fluid was carefully collected from the two experimental groups (Group 2 and Group 3–6) and fixed at a concentration of 1 ml. Cell death was analyzed by PI staining.
Cytokine analysis
To investigate the effect of PMIDA-CoO NPs on cytokine production, an ELISA was used for determination of TNF-α, IL-10 production of mouse serum from group 2 (tumor control) and 3–6 (tumor-induced PMIDA-CoO NPs treated at different concentration). All measurements were analyzed in triplicate.
Apoptotic markers in vitro
After treatment schedule, DL cells were collected and lysed and centrifuged to collect the supernatant. The supernatants were used for the detection of pro-apoptotic and anti-apoptotic markers using ELISA. All measurements were analyzed in triplicate.
Protein estimation
Protein was determined using bovine serum albumin as standard, according to Lowry et al. [26].
Statistical analysis
The data were expressed as mean ± SEM, n = 6. Comparisons between the means of control and treated group were made by one-way ANOVA test (using a statistical package, Origin 6.1, Northampton, MA 01060 USA) with multiple comparison t tests, p < 0.05 as a limit of significance.
Result and discussion
Magnetic NPs with a suitable surface coating show enhanced performance in biomedical applications compared to magnetic NPs [27]. One important approach is to modify the surface by coating with nontoxic materials such as PMIDA. We prepared PMIDA-CoO NPs by a thermal decomposition method. An acid-functionalized surface was generated by reacting with phosphonomethyl iminodiacetic acid. Based on this ability of phosphonic acid to exchange with the phosphate ions on crystals, PMIDA has been chosen as a robust anchor to functionalize the porous magnetic substrate with –COOH groups. The characterization and biological activities of the synthesized magnetic nanoparticles were by the following methods.
Characterization of NPs
X-ray diffraction (XRD)
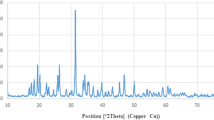
The crystal structure of synthesized PMIDA-CoO NP nanoparticles was determined by XRD. The diffractogram is given in Fig. 1a. The characteristic peaks at \(2\theta\) = 22.080, 36.490, 43.200, 52.600, 70.190 and 77.500 for cobalt oxide nanoparticles, which are marked respectively by their indices (111), (220), (311), (400), (511), and (440) in agreement with JCPDS card no 73-1701, appeared in both bare and PMIDA-CoO NPs. XRD results revealed the conjugation of PMIDA with CoO NPs did not affect the crystal formation of CoO NPs.

X-Ray diffraction study of PMIDA-coated cobalt oxide nanoparticles (a). DLS of PMIDA-coated cobalt oxide nanoparticles (b). TEM of PMIDA-coated cobalt oxide nanoparticles (c). SEM of PMIDA-coated cobalt oxide nanoparticles (d), and FT-IR spectra of PMIDA-CoO NPs (e)
DLS study
The measurement of the hydrodynamic size of PMIDA-CoO NPs in phosphate-buffered saline (PBS) by dynamic light scattering shows stable nonaggregated particles with a mean diameter of 80 ± 10 (Fig. 1b). The calculated size distribution histogram confirmed the size distribution of NPs (Fig. 1b). These NPs showed good stability in water.
Transmission electron microscopy and scanning electron microscopy
The TEM and SEM morphology of PMIDA-CoO NPs shows having nearly spherical geometry with a mean size of 62 ± 6 nm. The result was represented in Fig. 1c, d. The observed nanoparticle size was approximately larger than the hydrodynamic diameter obtained from the DLS experiment. TEM image showed the size in the dried state of the sample, whereas DLS measured the size in the hydrated state of the sample, so that the size measured by DLS was a hydrodynamic diameter and larger. However, one has to bear in mind that by TEM we image single particles, while DLS gives an average size estimation, which is biased toward the larger-size end of the population distribution. The size of the nanocomplex plays an important role in the delivery of anticancer agents, as the NPs up to ~400 nm can easily extravasate through the defective vasculature system in the tumor tissues and subsequently accumulate in the tumor microenvironment in presence of ineffective lymphatic clearance [28]. This process is known as “enhanced permeability and retention (EPR)” effect and forms the basis of “passive targeting” for in vivo delivery of anticancer drugs encapsulated in polymeric nanocarriers.
FTIR spectra show basic characteristics IR band of CoO at 566 cm−1 and a broad band around 3,436 cm−1 indicates the presence of –OH groups on the nanoparticle surface. After conjugation of PMIDA on the nanoparticle surface a significant decrease is seen in the intensity of the band at 3,436 cm−1 which indicates the conjugation of phosphonic acid on CoO NPs. A significant band also appears at 1,100, 1,740 cm−1 for M–O–P and P=O, respectively, which also confirms the successful conjugation of PMIDA on nanoparticle surface. The zeta potential of the PMIDA-CoO NPs was −33 mV and very stable in aqueous media. The dissolution assay (Fig. 2) shows the cobalt ion release pattern from PMIDA-CoO NPs in physiologic pH. Our result showed that PMIDA-CoO NPs release lower Co++ ion into the medium (U3—368 ppm and U4—86 ppm) than bare CoO NPs (U1—957 ppm and U2—522 ppm). The binding of PMIDA with CoO NPs reduces the Co++ ion release.

Cobalt ion release was measured by atomic absorption spectroscopy, where U1 and U2 denote Co++ ion release from bare cobalt (CoO) oxide nanoparticles and U3 and U4 denote Co++ ion release from PMIDA-CoO nanoparticles. Different concentrations of standards were used to determine the cobalt ion concentration
In vitro toxicity of PMIDA-CoO NPs on leukemic cells
PMIDA-coated cobalt oxide nanoparticles (PMIDA-CoO NPs) exhibit toxicity among the three cell lines (jurkat, KG1-A, and K562) tested which give promising activity against the used cells (Fig. 3). PMIDA-CoO NPs reveal toxicity against the jurkat cell line in a dose-dependent fashion. It was observed from our experiment that NPs kill the leukemia cell significantly at the dose of 10 µg/ml, 25 µg/ml and 50 µg/ml by jurkat cells by 28.83, 59.5, 67.58, KG-1A cells by 24.79, 54.8, 66.55, and K562 cells by 26.81, 58.79, 64.51 %, respectively (Fig. 3). Here the present findings demonstrate that cancerous white blood cells are markedly more susceptible to PMIDA-CoO NPs. The PMIDA-CoO NPs released higher amount of Co++ in cancerous environment and the Co++ induced toxicity to cancer cells. Our result correlates with the dissolution study. Bound with PMIDA, the surface- modified nanoparticles reduce the release of Co++ into the medium and induce lower toxicity to normal cells. But, the Co ion release was greater in cancer cell culturing medium because cancer cells secrete phospholipase D (PLD) which is essential for the breakage of P–O bonding of protein residues and helps in cancer metastasis [29–31] and PLD shows its optimum activity in between pH 5.0 and 6.0 [32]. In our study, we found a significant PLD level in the cancer supernatant (data not shown). The pH of the cancer microenvironment remains acidic (pH 5.0–6.0). The enzymatic cleavage (PLD2 mediated) of M–O–P bond from PMIDA-CoO NPs influenced the release of higher Co++ ions into cancerous medium and the ions were quickly internalized by cancer cells due to higher negative surface charge. Internalization of higher amount of Co++ ions altered the cellular balance and the cancer cells were died. From this point of view, we may hypothesize that phospholipase D may cleave the P–O bond of the PMIDA-CoO NPs and release Co++ into the medium and that Co++ ion kills the cancer cells.

Cytotoxicity of PMIDA-coated cobalt oxide nanoparticles against human leukemic cell lines. n = 6; values are expressed as mean ± SEM. Asterisk indicates the significant difference as compared to control group
In vitro toxicity studies on normal lymphocytes
The toxicity of the PMIDA-CoO NPs was checked toward normal human lymphocytes in vitro by MTT assay (Fig. 4). It was found that PMIDA-CoO NPs have little cytotoxic effects up to 25 µg/ml but at 50 µg/ml PMIDA-CoO NPs kill normal cells by 61.81 %. Hence the dose of 25 µg/ml surface-modified NPs is used for biomedical application. In this study, we examined the dose-dependent toxicity profiles of PMIDA-CoO NP to human primary immune cells. The cellular response is dynamic and the ultimate phenotype is affected by a myriad of competing or overlapping signals present in the microenvironment; studies were performed to determine how PMIDA-CoO NPs affect normal cells. Here the present findings demonstrate that PMIDA-CoO has a little effect on the normal counterparts of used cancer cells up to 25 µg/ml (Fig. 4). The toxicity of the PMIDA-CoO NPs depends on the Co++ ion release from nanocomplex. PMIDA is insoluble at physiological pH, maintained by normal cells and this was proved by dissolution studies which also reflect the release of the lower amount of Co++ into the media. The enzyme, phospholipase D was very low or may not present in normal cell and the enzymatic cleavage of the PMIDA-CoO NPs (P–O bond of PMIDA-CoO NPs) was arrested which inhibited the release of Co++ into the medium as well as intracellular system. Resultants, the low cytotoxicity was observed up to 25 µg/ml. But at higher concentration (50 µg/ml), the endocytosis of the NPs was more into cells and showed toxicity. Moreover, our results showed that PMIDA-CoO NPs released lower Co++ into the media and exerted lower toxicity to normal cells.

Cytotoxicity of PMIDA-coated cobalt oxide nanoparticles against normal human lymphocytes (a) and doxorubicin was used as control (b). n = 6; values are expressed as mean ± SEM. Asterisk indicates the significant difference as compared to control group
Cobalt ion release assay by atomic absorption spectroscopy (AAS)
We analyzed the supernatant after centrifugation of 25 µg/ml nanoparticles incubated in normal and cancer cell culture and supernatant was collected for Co ion concentration assay. Figure S1, S2 shows the Co2+ released from PMIDA-CoO NPs. Results showed that the PMIDA-CoO NPs released higher amount of Co++ ion (12.26, 11.64, 12.26 ppm) in cancer culture supernatant than in normal cell supernatant (4.69 ppm). This means that the cleavage of M–O–P bonding occurs in cancer environment.
Intracellular uptake of nanoparticles
The fluorescence images (Fig. 5a) shows the distribution of the PMIDA-CoO NPs in the cytoplasm leaving the clear zone of nucleus, indicating cellular uptake instead of adhering to the surface and the NPs preferentially target the cancer cells and were interned. This internalization might be due to the endocytosis [17, 33]. The toxicity in leukemia cells exposed to PMIDA-CoO NPs depends on the endocytosis of PMIDA-CoO NPs into the cell (Fig. 5a). Dysregulated cholesterol metabolism in leukemic cells [34] may enhance the uptake of PMIDA-CoO NPs into cancer cells. The higher internalization of the PMIDA-CoO NPs was confirmed by AAS study mentioned previously. In addition, tumor cell membranes contain higher amount of phospholipids than normal cells [35, 36]. The presence of high phospholipids may influence the attachment of the PMIDA-CoO NPs with cancer cell membrane, or the acids present in phospholipids may help to attach the PMIDA-CoO nanocomplex and release the free Co++ which enters into the cell and apoptosis by activation different biochemical process. It was established that the leukemic cells bear more negative charge on the cell membrane than normal lymphocytes, which may reduce the cellular internalizations of NPs. The positively charged PMIDA-CoO NPs were internalized more into cancer cells rather than normal cells. The negative–positive interaction happens in between cancer cell membrane and PMIDA-CoO NPs. The Co++ ion uptake was estimated by AAS. It was found that Co++ ion uptake was significantly elevated by cancer cells at 25 µg/ml doses after 8 h (0.52 pg Co++ ion/jurkat cell, 0.48 pg Co++ ion/KG-1Acell, 0.52 pg Co++ ion/K562 cell). On the other hand, no significant elevation of Co++ ion was noted in lymphocytes after 8 h (0.20 pg Co++ ion/cell) (Fig. 5b).Surface oxidation upon contact with cell culture medium or proteins in the cytoplasm liberates Co++ ions from cobalt-based NPs that could amplify the toxicity [26] whereas in PMIDA-CoO NP, the liberation of ions is less. The particle aggregation into the media is the vital issue of NPs. The results showed the time-dependent increase in concentration of Co++ ions. This ion liberation may be influenced by an enzyme PLD. Figure 5 shows that the concentration of free Co2+ ion/100 µl of culture media was slightly increased which suggests that no immediate effect was produced by the culture medium on the aggregation state of the NPs. The present study showed there were no such differences in the size, measured by DLS study, between the nanoparticles present in the culture medium after and before the incubation time (S3). These results suggest that no aggregation process is produced.

Cobalt ion concentrations in cancer cell and normal cell were measured by atomic absorption spectra. n = 6; values are expressed as mean ± SEM. Asterisk indicates the significant difference as compared to control group
PMIDA-CoO NP-induced ROS generation
The potential of PMIDA-CoO NPs to induce oxidative stress was assessed by measuring the ROS. Figure 6 shows that the PMIDA-CoO NPs significantly induced the intracellular production of ROS (p < 0.05) in cancer cells. NPs induced ROS up to 2.0-fold and 2.7-fold increase in jurkat cells, in KG-1A cells 1.92-, 2.44-fold and in K562 cells 1.87-, 2.61-fold at 25 and 50 µg/ml, respectively, from the negative control levels (Fig. 6A, B). A number of studies indicate that certain nanomaterials, including metal oxide NPs, have the potential to exhibit spontaneous ROS production based on material composition and surface characteristics while other nanomaterials trigger ROS production only in the presence of select cell systems [37–39]. As there is increasing evidence that elevated ROS acts as a critical signaling molecule in the induction of apoptosis induced by many different stimuli [40, 41], studies were performed to determine if an NP-induced cytotoxicity occurs via an apoptotic pathway. Results presented in Fig. 6A, B provide strong evidence that PMIDA-CoO NPs induce apoptosis in cancer cells with the production of ROS. Collectively, these studies indicate that a primary mechanism of PMIDA-CoO NPs induce cytotoxicity may proceed by inducing the generation of ROS which then underlies the induction of apoptosis.

Fluorescence micrograph of jurkat cell stained by DCFH2-DA (A), here a jurkat control, b jurkat + 10 µg/ml PMIDA cobalt oxide nanoparticles, c jurkat + 25 µg/ml PMIDA cobalt oxide nanoparticles, d jurkat + 100 µM H2O2, e KG-1A control, f KG-1A + 10 µg/ml PMIDA cobalt oxide nanoparticles, g KG-1A + 25 µg/ml PMIDA cobalt oxide nanoparticles, h KG-1A + 100 µM H2O2, i K562 control, j K562 + 10 µg/ml PMIDA cobalt oxide nanoparticles, k K562 + 25 µg/ml PMIDA cobalt oxide nanoparticles, l K562 + 100 µM H2O2. The fluorescence intensity of DCFH2-DA-stained jurkat cells was measured by spectrofluorimetry (B)
Role of NAC in NP-mediated cytotoxicity
Experiments were performed to determine if the cancer cell death that results from NP exposure is dependent on the generation of intracellular ROS. Jurkat cells were exposed to increasing concentrations of the ROS quencher, NAC (N-acetyl cysteine) [42, 43]. NAC is a cysteine derivative compound and has a biologically important organic substance that is known to have metal-chelating properties and can be used for metal poisoning therapy [51, 52]. The six-membered ring of NAC reacts with metal ions via the thiol and carboxyl groups and chelated them. The intracellular Co++ ion released from the PMIDA-CoO NPs into the cancer cells may be chelated by NAC, in this way, NAC may reduce the level of intracellular ROS generation [52, 53]. Figure 7 shows that NAC has significant effects to prevent NP-induced cytotoxicity with rescue being observed at different concentrations of NP tested. Significant differences (p < 0.05) were observed between cultures not pre-treated with NAC and both NAC pretreatment (10 mM) for each NP concentration tested. For example, with 10 mM NAC, nearly 100 % viability was retained even at an NP concentration previously shown to reduce cell viability below 10 %. These results indicate that ROS generation plays a causal role in NP-induced cytotoxicity. The apoptosis is prevented by the addition of ROS quencher at the dose of 5 mM and 10 mM concentration. These observations provide the basis for the NP- mediated apoptosis. The development of free radicals is directly responsible for DNA strand breaks or disruption of DNA [44].

Cytotoxicity of PMIDA-coated cobalt oxide nanoparticles against jurkat, KG-1A and K562 cell lines with NAC per-treatment at 10 mM concentration (n = 6); values are expressed as mean ± SEM. Asterisk indicates the significant difference as compared to control group
DNA fragmentation
The genotoxic effects of PMIDA-CoO NPs were determined using agarose gel. After electrophoresis, nanoparticle-treated cells were examined under BioRad Gel Doc and images were captured. The DNA fragmentation was observed in leukemic cells treated with 25 µg/ml (Fig. 8). It was observed that PMIDA-CoO NPs induced DNA damage in jurkat, KG-1A and K562 cell lines. At the highest concentration of nanoparticles (25 µg/ml), DNA damage was increased.

DNA fragmentation assay of PMIDA-coated cobalt oxide nanoparticles on jurkat (1 control, 2 treated with 25 µg/ml), KG-1A (3 treated with 25 µg/ml, 4 control), and K562 cells (5 treated with 25 µg/ml, 6 control)
Apoptosis or necrosis study
PMIDA-CoO NP-induced apoptosis or necrosis was examined in leukemic cells using acridine orange and ethidium bromide dual staining visualized under fluorescence microscope. Viable cells took the green color of acridine orange with normal cell morphology. Acridine orange- and ethidium bromide-stained cells with condensed nucleus are in early and late apoptosis stages, and red color cells were necrotic cells, respectively. The total number and percentage of leukemic cells with ethidium bromide staining emitting red fluorescence increased (Fig. 9). The result showed that PMIDA-CoO NPs induces apoptosis followed by necrosis to leukemic cells depending on the dose of NPs and incubation time.

Images of AO/EB dual staining the non-treated 25, 50 μg/ml of PMIDA-CoO NP-treated jurkat, KG1-A, and K562 cells after 12 h of treatment. Condensed chromatin in early apoptotic (green) cells and fragmented chromatin in late apoptotic (red/reddish) cells are clearly visible
Apoptotic markers
The concentrations of pro-apoptotic markers from NP-treated cells enlighten the cell death pathway. Higher concentration of caspase-8 and caspase-3 indicates the extrinsic pathway of apoptosis. The higher release of p38 supports the apoptosis pathway (Fig. 10). The immunofluorescence (immunocytochemistry) study showed that the activation of caspase-8, caspase-3 and p38 in higher amount leads to the apoptosis of the leukemic cells (Fig. 11). Oxidative stress triggers various signaling pathways, including mitogen-activated protein kinase (MAPK) cascades and p38 MAPK, were shown to be key mediators of stress signals [50]. The activation of p38 mitogen-activated protein kinase (MAPK) upregulates caspase activation-induced apoptosis. Results showed the activation of p38 MAPK, caspase-8 and caspase-3. The generation of ROS may induce p38 MAPK, caspase-8 and caspase-3; this generation of reactive oxygen species is directly involved with PMIDA-CoO NP-mediated cell death by the activation of the p38 pathway. The FACS analysis also supports our results (Fig. 12).

Alteration of pro- and anti-apoptotic response of PMIDA-coated cobalt oxide nanoparticles on jurkat cell (a), KG-1A cell (b), K562 cell (c). Caspase-8, caspase-3, p38, pAKT, BCL-2; (n = 6) values are expressed as mean ± SEM. Asterisk indicates the significant difference as compared to control group

Leukemic cells treated with PMIDA-CoO NPs at 25 μg/ml for 24 h. The induction of apoptosis was estimated by the expression of caspase-8, caspase-3 and p38 proteins were detected by immunofluorescence. Here a jurkat control, b jurkat caspase-8, c KG-1A control, d KG-1A caspase-8, e K562 control, f 562 caspase-8; g jurkat control, h jurkat caspase-3, i KG-1A control, j KG-1A caspase-3, k K562 control, l K562 caspase-3; m jurkat control, n jurkat p38, o KG-1A control, p KG-1A p38, q K562 control, r K562 p38

The expression of caspase-8 and caspase-3 (a) and pAKT, BCL-2 (b) in leukemic cells treated with PMIDA-coated cobalt oxide nanoparticles were measured by FACS. The NP-treated cells were permeabilized and then treated with anti-caspase-8, caspase-3, anti-p3, pAKT, BCL-2. After 1-h incubation, the cells were collected by sequential washing and allowed to incubate with anti-human antibody tagged with Texas Red and FITC. The stained cells were examined by FACScallibure and the data were interpreted using CellQuest software
Cytokine analysis
After the treatment schedule, cell-free supernatants were used to quantify cytokine levels using an ELISA assay. The results demonstrate significant dose-dependent increases in TNF-α at all NP concentrations tested (Fig. 13). The results showed that the leukemic cells are producing high levels of TNF-α. However, since exposure to particles induced TNF-α increases up to 2.43-fold in jurkat, 1.86-fold in KG1-A, and 1.88-fold in K562 at 25 µg/ml PMIDA-CoO NPs (Fig. 10). It is likely that leukemic cells are produced TNF-α in response to PMIDA-CoO particle exposure. The anti-inflammatory cytokine IL-10 reduces up to 2.76-fold in jurkat, 2.71-fold in KG1-A, and 2.5-fold in K562 at 25 µg/ml PMIDA-CoO NPs (Fig. 13). PMIDA-CoO NP induces pro-inflammatory cytokines in parallel with the deduction of the anti-inflammatory cytokines. Studies were performed to evaluate the ability of PMIDA-CoO NPs to modulate TNF-α cytokine production in primary human immune cells. These particular cytokines were chosen because they represent critical pathways involved in the inflammatory response and differentiation processes. The results demonstrate significant dose-dependent increases in TNF-α at all NP concentrations tested. These results suggest that a synergistic relationship between PMIDA-CoO NPs and TNF-α may occur in in vitro settings employing PMIDA-CoO NPs, and demonstrate that PMIDA-CoO NPs were capable of inducing at least some key components of inflammation. The ability of PMIDA-CoO NPs to induce pro-inflammatory cytokine expression in human primary immune cells is consistent with the recognized relationship between oxidative stress and inflammation. PMIDA-CoO NPs induce high levels of TNF-α help to promote Th1 differentiation [45–47] as well as functioning as a regulator of acute inflammation [47]. Thus, our findings indicate that careful titration of PMIDA-CoO NP-based therapeutic interventions may be successful in elevating a group of cytokines important for eliciting a Th1-mediated immune response with effective anticancer actions. However, high level and/or chronic exposure to TNF-α has been shown to produce serious detrimental effects on the host, including septic shock or symptoms associated with autoimmune disease [47]. Our results demonstrate significant dose-dependent increases in TNF-α over a somewhat narrow range of PMIDA-CoO NP concentrations. The magnitude of TNF-α induction, as well as other pro-inflammatory cytokines, and their local, regional delivery to tumor sites or other desired areas, will undoubtedly be important parameters when considering PMIDA-CoO NPs for biomedical purposes to achieve the desired therapeutic response without eliciting potential systemic damaging effects from these cytokines.

Pro- and anti-inflammatory response of PMIDA-coated cobalt oxide nanoparticles on jurkat, KG-1A and K562 cells. TNF-α, IL-10 (a); (n = 6) values are expressed as mean ± SEM. Asterisk indicates the significant difference as compared to control group
Effect of PMIDA-CoO NPs on tumor volume and body weight
Treatment with PMIDA-CoO NPs for a period of 15 days in DLA tumor-bearing mice led to a significant reduction in tumor volume in comparison with tumor controls. Administration of PMIDA-CoO NPs extended the life span of tumor-bearing mice. From the first day of tumor induction we found that the untreated tumor control mice were survived for only 17 days which was extended up to 29 days for CoO NP-treated tumor-bearing mice (Fig. 6a). Tumor volume in control mice was about 8.18 ml but was significantly reduced to 2.76 ml in the group treated with PMIDA-CoO NPs at a concentration of 1,000 µg/kg BW for 15 days. Body weight, measured throughout the period of the experiment, was reduced in the treated tumor-bearing group when compared with the tumor control group (Fig. 14b). Figure 14b, c shows the confirmation of the antitumor activity of PMIDA-CoO NPs by significant reduction in tumor volume by killing of cancer cells in the IP region in comparison with the tumor control mice, thereby serving the tumor mice to regain its original weight.

DLA-infected mice treated with PMIDA-CoO NPs at 100, 200, 500 and 1,000 μg/kg BW in PBS via intraperitoneal (IP) injection for 15 (a) survival time, (b) tumor volume, (c) cytotoxicity of DLA cells, (d) pro-anti-inflammatory response, (e) pro- and anti-apoptotic response of PMIDA-coated cobalt oxide nanoparticles’ treated, tumor control and normal mice; (n = 6) values are expressed as mean ± SEM. Asterisk indicates the significant difference as compared to control group. (f) Generation of ROS in PMIDA-CoO NP-treated cancer cells (DLA)
Viability analysis of DLA cells
Viability analysis of DLA cells of peritoneal fluid revealed that PMIDA-CoO NPs treatment in tumor-bearing mice led to a significant reduction in the number of malignant cells in the treated group when compared with the control group (Fig. 14c), reflecting the potential of PMIDA-CoO NPs to have cytotoxic effects on tumor cells, without affecting normal cells. The DL cells produce a significant amount of ROS after completion of NPs treatment (Fig. 14f). The in vivo study showed a dose-dependent effect of CoO NPs on DLA cell lines assessed by MTT assay. The PMIDA-CoO NPs induced significant reduction in cell viability in comparison with controls. The cytotoxic effect of PMIDA-CoO NPs on cell viability plays a major role in antitumor activity. In the present study, IP inoculation of DLA cells in mice produced a marked increase in the cancer cell count, which indicated tumor progression in the animals, whereas a substantial decrease in cancer cell numbers in the PMIDA-CoO NP-treated tumor-bearing mice was observed through cytopathologic analysis. Treatment with PMIDA-CoO NPs clearly showed significant inhibitory effects on tumor cell proliferation and survival. The effect of PMIDA-CoO NPs in increasing mean survival time and lifespan depends on their ability to reduce tumor cell viability and induce cytotoxicity. The role of CoO NPs in inhibiting three different leukemic cells in vitro and DLA cell proliferation in vivo was established the potentiality of nanoparticles against cancer treatment.
Cytokines
In vivo cytokine release and activation of pro-apoptotic markers proved the in vivo anticancer activity of PMIDA-CoO NPs. We found that PMIDA-CoO NP treatment increases the TNF-α level and decreases the IL-10 level in tumor-bearing mice, whereas the cytokine scenario was vice versa in tumor control mice (Fig. 14d). The increasing concentration of TNF-α was helping to increase the caspase-8 and provoke the extrinsic pathway of apoptosis (Fig. 14e). The PMIDA-CoO NPs serve as antitumor agents by increasing pro-apoptotic factors and decreasing anti-apoptotic factors. Taken together, our data suggest that PMIDA-CoO NPs can induce cytotoxic effects on DLA cells, inhibiting tumor progression and thereby effectively controlling tumor progression.
From the results obtained in this study, we suggest that TNF-α-induced signaling was one of the responsible factors for NP- mediated cell death.
Conclusions
In conclusion, the above study indicated the effective use of PMIDA-modified CoO NPs on human leukemic cell lines (jurkat, K562 and KG1A cells). Our results showed that exposure of leukemic cell lines to surface-modified CoO NPs caused reactive oxygen species (ROS) generation caused DNA damage in leukemic cell lines was reflected by an increase in cell death of jurkat, KG-1A and K562 cells. Surface-modified CoO NPs induced cell death by increasing the release of Co++ ions that depends on the activity of phospholipase D2 in cancer microenvironment. The enzymatic cleavage of M–O–P bond from PMIDA-CoO NPs influenced the release of higher Co++ ions into cancerous medium and the ions were quickly internalized by cancer cells due to higher negative surface charge. Internalization of higher amount of Co++ ions induced higher amount of ROS in association with TNF-α and p38 MAPK. Oxidative stress can trigger the activation of p38 MAPK signaling pathways that influence the cytotoxicity observed in affected cells. The activation of ROS and TNF-α may be involved in the surface-modified CoO NP-induced leukemic cell death since the inhibition of either pathway resulted in attenuation of apoptosis response. The in vivo study shows that PMIDA-CoO NPs were efficiently killed DLA cells. These findings have important implications for understanding the potential health effects of metal nanoparticles exposure.
References
Lee JH, Huh YM, Jun YW et al (2007) Nat Med 13:95–99
Rana S, Gallo A, Srivastava RS, Misra RK (2007) Acta Biomat 3(2):233–242
Sun C, Lee JS, Zhang M (2008) Adv Drug Deliv Rev 60(11):1252–1265
Chertok B, Moffat BA, David AE et al (2008) Biomaterials 29(4):487–496
Ahamed M, Karns M, Goodson M et al (2008) Toxicol Appl Pharmacol 233(3):404–410
Wang K, Xu JJ, Chen HY (2005) Biosens Bioelectron 20:1388–1396
Liu X, Qiu G, Li X (2005) Nanotechnology 16:3035–3040
Parkes LM, Hodgson R, Lu LT, Tung LD, Robinson I, Fernig DG, Thanh NT (2008) Contrast Media Mol Imag 3:150–156
Papis E, Rossi F, Raspanti M, Isabella DD, Colombo G, Milzani A, Bernardini G, Gornati R (2009) Toxicol Lett 189:253–259
Dailey JP, Phillips JP, Li C, Riffle JS (1999) J Magn Magn Mater 194:140–148
Rutnakornpituk M, Baranauskas V, Riffle JS, Connolly J, Pierre TG, Dailey JP (2002) Eur Cells Mater 3:102–105
Pardoe H, Chua-anusorn W, Pierre TG, Dobson J (2001) J Magn Magn Mater 225:41–46
Hubert PM, Guerrero G, Vioux A (2005) J Mater Chem 15:3761–3768
Adden N, Gamble LJ, Castner DG, Hoffmann A, Gross G, Menzel H (2006) Langmuir 22:8197
Neouze MA, Schubert U (2008) Monatsh Chem 139:183–195
Ghosh T, Chattopadhyay T, Das S, Mondal S, Suresh E, Zangrando E, Das D (2011) Cryst Growth Des 11:3198–3205
Mohapatra S, Mallick SK, Maiti TK, Ghosh SK, Pramanik P (2007) Nanotechnology 18:385102–385111
Hudson L, Hay FC (1991) Practical immunology, 3rd edn. Blackwell scientific publications, Oxford/London/Edinburgh/Boston/Melbourne, pp 21–22
Chattopadhyay S, Dash SK, Ghosh T, Das D, Pramanik P, Roy S (2013) Cancer Nano 4:103–116
Gaither LA, Eide DJ (2001) J Biol Chem 276(25):22258–22264
Hanley C, Layne J, Punnoose A, Reddy KM, Coombs I, Coombs A, Feris K, Wingett D (2008) Nanotechnology 19:295103
Zhou BR, Marina G, Marina F et al (2003) Acta Pharmacol 24:193–198
Sheikpranbabu S, Kalishwaralal K, Lee KJ, Vaidyanathan R, Eom SH, Gurunathan S (2010) Biomaterials 31:1318–1329
Kalimuthu K, Pandian SRK, Deepak V, Bilal M, Gurunathan S (2008) Colloid Surf B Biointerfaces 65:150–153
Sriram M, Kanth S, Kalishwaralal K, Gurunathan S (2010) Int J Nanomed 5:753–762
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) J Biol Chem 193:265–275
Das M, Mishra D, Maiti TK, Basak A, Pramanik P (2008) Nanotechnology 19:415101
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R (2007) Nat Nanotechnol 2:751
Sant S, Poulin S, Hildgen P (2008) J Biomed Mater Res A 87:885–895
Foster AD, Xu L (2003) Mol Can Res 1:789–800
Barth A, Bezlyepkina N (2004) J Biol Chem 279:51888–51896
Quarles RH, Dawson RMC (1969) Bio chem J 112:795
Lovric J, Cho SJ, Winnik FM, Maysinger D (2005) Chem Biol 12:1227–1234
Resnitzky P, Bustan A, Peled A, Marikovsky Y (1988) Leuk Res 12:315–320
Sanpui P, Chattopadhyay A, Ghosh SS (2011) ACS Appl Mater Interfaces 3:218–228
Bergelson LD, Dyatlovitskaya EV, Sorokina IB, Gorkova NP (1974) Biochim Biophys Acta 360(3):361–365
Xia T, Kovochich M, Brant J, Hotze M, Sempf J, Oberley T, Sioutas C, Yeh JI, Wiesner MR, Nel AE (2006) Nano Lett 6:1794–1807
Long TC, Saleh N, Tilton RD, Lowry GV, Veronesi B (2006) Environ Sci Technol 40:4346–4352
Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM (2007) Antioxid Redox Signal 9:49–89
Carmody RJ, Cotter TG (2001) Redox Rep 6:77–90
Yang MH, Jiang JH, Yang YH, Chen XH, Shen GL, Yu RQ (2006) Biosens Bioelectron 21:1791–1797
Boudreau RT, Conrad DM, Hoskin DW (2007) Exp Mol Pathol 83:347–356
Gupta AK, Gupta M (2005) Biomaterials 26:3992–4021
Ishikawa K, Ishii H, Saito T (2006) DNA Cell Biol 25:406–411
Dong C, Flavell RA (2001) Curr Opin Hematol 8:47
Croft M (2009) Nat Rev Immunol 9:271
Fishman MA, Perelson AS (1999) Bull Math Biol 61:403
Joshi SG, Sahni SK (2003) Histochem Cell Biol 119:463–468
Fernandes NV, Guntipalli PK, Huanbiao MO (2010) Anticancer Res 30:4937–4944
Hibi M, Lin A, Smeal T, Minden A, Karin M (1993) Genes Dev 7:2135–2148
Chattopadhyay S, Chakraborty SP, Laha D, Baral R, Pramanik P, Roy S (2012) Cancer Nano 3:13–23
Luczak MW, Zhitkovich A (2013) Free Radic Biol Med 65:262–269
Guzeloglu S, Yalcin G, Pekin M (1998) J Organomet Chem 568:143–147
Xu G, Ahn J, Chang S, Eguchi M, Ogier A, Han S, Park Y, Shim C, Jang Y, Yang B, Xu A, Wang Y, Sweeney G (2012) J Biol Chem 287:4808–4817
Armstrong JS, Steinauer KK, Hornung B, Irish JM, Lecane P, Birrell GW, Peehl DM, Knox SJ (2009) Cell Death Differ 9:252–263
Azaria LH, Kirkpatrick CJ, Korenstein R, Marche PN, Maimon O, Ponti J, Romano R, Rossi F, Schindler UG, Sommer D, Uboldi C, Unger RE, Villiers C (2011) Toxicolog Sci 122:489–501
Colognato R, Bonelli A, Ponti J, Farina M, Bergamaschi E, Sabbioni E (2008) Mutagenesis 23:377–382
Ponti J, Sabbioni E, Munaro B, Broggi F, Marmorato P, Franchini F (2009) Mutagenesis 24:439–445
Petrarca C, Perrone A, Verna N, Verginelli F, Ponti J, Sabbioni E (2006) Int J Immunopathol Pharmacol 19:11–14
Levard C, Mitra S, Yang T, Jew AD, Badireddy AR, Lowry G, Brown G (2013) Environ Sci Technol 47:5738–5745
Hernandez-Flores G, Ortiz-Lazareno PC, Lerma-Diaz JM, Dominguez-Rodriguez JR, Jave-Suarez LF, Aguilar-Lemarroy AC, de Celis-Carrillo R, del Toro-Arreola S, Castellanos-Esparza YC, Bravo-Cuellar A (2011) BMC Cancer 11:483
Acknowledgments
The authors express gratefulness to the Department of Biotechnology, Government of India for funding. The authors also express gratefulness to Indian Institute of Technology, Kharagpur and Vidyasagar University, Midnapore for providing the facilities to execute these studies.
Conflict of interest
Authors declare that there are no conflicts of interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chattopadhyay, S., Dash, S.K., Tripathy, S. et al. Phosphonomethyl iminodiacetic acid-conjugated cobalt oxide nanoparticles liberate Co++ ion-induced stress associated activation of TNF-α/p38 MAPK/caspase 8-caspase 3 signaling in human leukemia cells. J Biol Inorg Chem 20, 123–141 (2015). https://doi.org/10.1007/s00775-014-1221-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-014-1221-7