
Abstract
In addition to its electron transfer activity, cytochrome c is now known to trigger apoptosis via peroxidase activity. This new function is related to a structural modification of the cytochrome upon association with anionic lipids, particularly cardiolipin present in the mitochondrial membrane. However, the exact nature of the non-native state induced by this interaction remains an active subject of debate. In this work, using human cytochromes c (native and two single-histidine mutants and the corresponding double mutant) and micelles as a hydrophobic medium, we succeeded, through UV–visible spectroscopy, circular dichroism spectroscopy and NMR spectroscopy, in fully characterizing the nature of the sixth ligand replacing the native methionine. Furthermore, careful pH titrations permitted the identification of the amino acids involved in the iron binding over a range of pH values. Replacement of the methionine by lysine was only observed at pH above 8.5, whereas histidine binding is dependent on both pH and micelle concentration. The pH variation range for histidine protonation is relatively narrow and is consistent with the mitochondrial intermembrane pH changes occurring during apoptosis. These results allow us to rule out lysine as the sixth ligand at pH values close to neutrality and reinforce the role of histidines (preferentially His33 vs. His26) as the main candidate to replace methionine in the non-native cytochrome c. Finally, on the basis of these results and molecular dynamics simulations, we propose a 3D model for non-native cytochrome c in a micellar environment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cytochrome c is a soluble, small and basic haemoprotein that is located in the mitochondrial intermembrane space. Its historic function is to shuttle electrons from cytochrome c reductase to cytochrome c oxidase in the respiratory chain. In the native state, His18 and Met80 are the two axial ligands forming the iron hexacoordinated low-spin state (LSHM). However, cytochrome c has been shown to be deeply involved in the early steps of apoptosis [1]. Electrostatic and hydrophobic interactions with cardiolipin, an exclusive mitochondrial phospholipid, are at the origin of the destabilization of the haem iron–sulphur bond (Fe–Met80), leading to non-native low-spin species and high-spin pentacoordinated cytochrome c (for a recent review, see Kagan et al. [2]). In the high-spin form, haem iron can bind H2O2 and acquires peroxidase activity [3], generating cardiolipin hydroperoxide, which has been shown to be a key stage preceding release of cytochrome c from the mitochondria to the cytosol [4]. In the cytosol, cytochrome c interacts with Apaf-1 to form the apoptosome, a protein complex that activates the caspase cascade leading to apoptosis [5]. The mechanism by which the cytochrome c function is switched from an electron carrier to a peroxidase has not been fully elucidated. This new property of cytochrome c can be used to develop new anticancer drugs. Classically, anticancer treatments (radiotherapy and chemotherapy) create cellular stress, inducing escape of cytochrome c from the mitochondria and leading to the death of immune cells. So, latency periods between two treatment sessions are necessary to recreate the patient’s immune cell pool. Therefore, in this context, improvement of stimulation of apoptosis in tumour cells or inhibition of apoptosis in normal cells may be new effective strategies [6]. Atkinson et al. [7] have focussed on the inhibition of cytochrome c/cardiolipin peroxidase activity by different ligands that are able to bind to the haem iron of cytochrome c upon its structural modification by lipid interactions, but a better understanding of the structural modifications of cytochrome c in hydrophobic media is needed.
Despite numerous studies, the exact nature of the structural modifications of cytochrome c during interaction with the inner mitochondrial membrane remains unclear. Differences in the membrane model systems as well as the techniques and experimental conditions can yield different conclusions. For example, lipid specificity has been reported to correspond to different levels of non-native states of cytochrome c [8, 9] or depending on the experimental pH, in a micellar medium, histidine [10] and lysine [11] have been proposed as the ligand replacing the native methionine. The lipidic interaction of cytochrome c is believed to be both electrostatic and hydrophobic, inducing methionine decoordination and the formation of non-native low-spin states and a high-spin cytochrome c [9, 12–16]. The latter form is believed to be involved in the peroxidation of the acyl side chains of cardiolipin. Sodium dodecyl sulphate (SDS) has been used as an appropriate hydrophobic medium because its is an adequately spectroscopically silent model, permitting the use of various techniques. SDS micelles are particularly well adapted for NMR studies because of the fast tumbling of the vesicles and the availability of deuterated molecules. Furthermore, their low turbidity—compared with phospholipid vesicles—allows us to use large micelle to cytochrome c ratios without any saturation of the absorbance in UV–visible spectroscopy or circular dichroism (CD) spectroscopy. The addition of micelles has been shown to mimic the interaction of cytochrome c with anionic lipids displaying both electrostatic and hydrophobic interactions. At submicellar SDS concentration, the interaction is mainly hydrophobic, whereas SDS at micellar concentration increases the surface charge density and promotes the loss of stable tertiary structure through electrostatic interactions [17]. With use of SDS, methionine decoordination and the formation of non-native low-spin and high-spin species have been reported [10, 17–25], as has the peroxidase activity induced by cardiolipin [26]. In the presence of SDS, cytochrome c adopts a molten globule state, suggesting that its tertiary structure is profoundly perturbed but most of the secondary structure elements are preserved [27] as observed with phospholipidic vesicles [28, 29]. Nevertheless, the high-spin content present at high SDS concentration was shown to be species-dependent. In the case of yeast cytochrome c, more non-native low-spin species are present, in contrast to what is found for horse heart cytochrome c [27].
In our work, human cytochrome c was used to better understand the structural modifications that occur in the presence of SDS. Although the decoordination of Met80 is well documented, the non-native low-spin cytochrome c species, an intermediate between the native cytochrome c and the high-spin cytochrome c, is still a subject of debate. Different amino acids have been proposed to replace the methionine as the sixth axial ligand in the presence of cardiolipin or micelles. Whereas His26 or His33 has been proposed to play this role in the presence of lipids or micelles [10, 22, 24] or under denaturing conditions [30, 31], lysines have also been suggested to replace Met80 [32–35].
To study these non-native low-spin species, we designed and produced different mutants of human cytochrome c: H26Q, H33N and H26Q/H33N. SDS micelles were used to produce a hydrophobic environment that mimics the interaction of cytochrome c with phospholipids [10, 17, 22–25]. SDS titrations of cytochrome c solutions at different pH values were performed using different spectroscopic methods: NMR spectroscopy, CD spectroscopy and UV–visible spectroscopy. Changes in the coordination spin state modify the electronic environment of iron and its spectral characteristics, which have been analysed to fully describe the influence of SDS on cytochrome c. With use of an interactive molecular simulation approach, these experimental results allowed us to create an original model of the conformational changes of micellar cytochrome c. Finally, these results allow us to reconcile the ambiguities concerning the nature of the sixth ligand by taking into account the role of pH as well as micelle concentration.
Materials and methods
Materials
SDS (98.5 %) and horse heart cytochrome c were acquired from Sigma (St Louis, USA) and were used without any further purification. SDS-d 25 was purchased from Cortecnet (Voisins-le-Bretonneux, France).
Human cytochrome c mutations, expression and purification
Full details of the preparation of mutated proteins can be found in the electronic supplementary material. The plasmid pET21-CCHL-HuCytc was generously provided by Jeng et al. [36]. Human cytochrome c was expressed and purified as previously reported [36, 37].
Absorbance measurement
Absorption spectra in the Q band region (480–800 nm) and in the Soret band region (280–480 nm) were obtained with samples containing 20 μM ferricytochrome c (wild type, H26Q, H33N and H26Q/H33N) in phosphate buffer (50 mM, pH 6.8). Titrations were performed by adding small volumes (1 or 2 μl) of SDS solution to obtain appropriate concentrations ranging from 0 to 6 mM. Spectra were recorded at room temperature with a JASCO V-650 spectrophotometer with 10 mm path length quartz cells.
Circular dichroism
CD spectra in the near-UV region were recorded with a JASCO J-815 CD spectrophotometer; spectra were obtained with the same samples used for absorbance measurement with 10 mm path length cells. Measurements were recorded with a bandwidth of 1 nm, a scanning speed of 100 nm/min and a data pitch of 0.5 nm. All spectra were measured against an appropriate reference, with two scans being accumulated for each sample. The cell holder was thermostated with a circulating water bath (JASCO PTC-4235/15), and all spectra were recorded at 293.1 ± 0.1 K.
NMR spectroscopy
All spectra were recorded at 313 or 323 K using a Bruker Avance 500 spectrometer equipped with a triple-resonance TXI cryoprobe (1H, 13C and 15N). Spectra were acquired in deuterated solutions, and pH values were obtained by subtracting 0.4 U from the pH meter reading. 1H NMR spectra were recorded with a 90° reading pulse with a large window (100 kHz) to detect the cytochrome c paramagnetic signals and a small window of 6 kHz corresponding to the classic diamagnetic region. An acquisition time of 80 ms and a repetition time of 0.5 s were used for the large window, whereas 0.8 and 1.3 s were used for the small window. Heteronuclear experiments were conducted with natural abundance cytochrome c at a concentration ranging from 0.1 to 1 mM. With use of standard Bruker sequences, heteronuclear single-quantum correlation (HSQC) spectra were recorded in phase-sensitive mode in both dimensions with echo–antiecho time proportional phase incrementation gradient selection. 1H–13C HSQC spectra were acquired with 96 transients, spectral windows of 16 ppm/40 ppm in the proton/carbon dimensions and the carrier set at the water frequency and 130 ppm. Magnetization transfers were optimized for a coupling constant of 200 Hz, as previously described [38]. No carbon decoupling was applied during acquisition. A relaxation delay of 1 s was used, and a matrix of 2,048 × 96 points was acquired and transformed to 2,048 × 512 points after shifted sine square multiplication.
Molecular modelling
The solution structure of human cytochrome c proposed by Jeng et al. [36] (Protein Data Bank code 1J3S) was used as the simulation starting point for the native six-ligand state. The bishistidine state was built by interactive molecular dynamics performed on the native state [39]. Steered positioning of both histidines in an axial trans orientation was obtained by a rearrangement of the very flexible loop (residues 20–37), which induced an N-terminal helix rotation. This motion allowed the haem group to tilt with a minimal energy cost. The protein structures were described by parameters of the AMBER-derived force field of the program YASARA [40] and were placed in a neutralized (0.9 % NaCl) TIP3P solvent box, the single bishistidine low-spin form (LSHH) being inserted into the SDS micelle. Haem c parameters were defined after an automated AutoSMILES procedure (AM1 MOPAC calculation followed by a restrained electrostatic potential charge-fitting). The iron coordination geometry was reinforced upon simulation using distance-based harmonic restraints in accordance with the experimental NMR structure chosen as the starting point (model 1, Protein Data Bank code 1J3S) [36]. Both systems (native LSHM and non-native LSHH) were simulated under periodic boundary conditions and in the NVT ensemble (298 K). Structures were relaxed by successive steepest descent energy minimization followed by short molecular dynamics simulation (1 ns), providing a stable root mean square deviation over the last simulation steps. Production periods of 20 ns were collected every 2 ps. The final structural models correspond to the minimized most representative structure of the most populated cluster (Cα root mean square deviation less than 1.0 Å) found along the whole trajectory.
Results
Definition of haem coordination and spin-state markers
The three known cytochrome c spin states are as follows: the six-coordinated LSHM form, in which His18 and Met80 are the axial ligands; the non-native low-spin six-coordinated configuration in which Met80 is replaced by another strong-field axial ligand such as histidine or lysine (LSHX); and the high-spin state.
Two optical spectroscopic techniques, UV–visible spectroscopy and CD spectroscopy, were used to follow these changes. Figure S1 shows the reference spectra of the three main reported cytochrome c forms [20], namely the native form, the non-native low-spin form obtained at intermediate SDS concentration and the high-spin form observed in the presence of high SDS concentration. In the 480–800-nm region, the 695-nm band is indicative of the native state with Met80 haem coordination (LSHM) [41], whereas the band at 625 nm is characteristic of a high-spin configuration [42]. Thus, the absence of the 625- and 695-nm bands corresponds to one non-native low-spin state of cytochrome c (LSHX). In the Soret band region (350–450 nm), the maximum intensity at 409 nm is indicative of native ferricytochrome c (LSHM). A blue shift of the Soret band is observed for both the non-native low-spin state, with a Soret band maximum at 408 nm, and the high-spin form, in the range from 397 to 403 nm, depending on the experimental conditions.
The near-UV CD spectra (250–450 nm) reflect changes in the tertiary structure that affect the environment of aromatic protein side chains. The optical activity in the Soret band region is generated through the coupling of haem π–π* electric dipole transition moments with aromatic residues of the protein [43]. A strong Cotton effect is observed for the native form, with a negative band at 415 nm and a positive band at 398 nm. By contrast, a single sharp intense, positive band at 409 nm is characteristic of the non-native low-spin configuration. The high-spin form displays a broad positive signal centred at 403 nm.
Comparison of human wild-type and H26Q/H33N double-mutant cytochrome c below the critical micelle concentration of SDS
Titration below the critical micelle concentration (cmc) of SDS was performed at pH 6.8 for both the wild-type form and the H26Q/H33N double-mutant form of human cytochrome c (Fig. 1). In the absence of SDS, the spectra of the mutant and wild-type proteins are very similar, with the characteristic charge transfer band at 695 nm characteristic of the iron–methionine bond. For wild-type cytochrome c, the disappearance of the native low-spin form is complete at a 40:1 SDS to cytochrome c ratio. A strong positive band at 409 nm in the CD spectra and a Soret band at 408 nm are good markers of the non-native low-spin species (Fig. 1a, b). By contrast, addition of SDS below the cmc to the H26Q/H33N double mutant yields a different result. The titration was followed by UV–visible spectroscopy (Fig. 1c). The addition of SDS induces a decrease in the intensity of the charge transfer band at 695 nm, whereas a Q band and a Soret band at 625 and 402 nm, respectively, appear concomitantly. The interconversion of the two forms is further confirmed by the presence of isosbestic points during the titration. The CD absorption spectra of the double-mutant protein are also very informative. Starting with the classical Cotton effect in the absence of micelles, the addition of SDS leads to the loss of the Cotton effect and a weakly shifted broad positive band (Fig. 1d) that was previously associated with the high-spin form [43]. Thus, at these micelle-to-protein ratios, the absence of any non-native low-spin form, in contrast to the wild-type protein, demonstrates indirectly that His26 or His33 (vide infra) is involved in the LSHX form of wild-type cytochrome c at pH 6.8.

Visible absorption spectra (left) and near-UV circular dichroism spectra (right) of wild-type (a, b) and H26Q/H33N (c, d) human cytochrome c at different sodium dodecyl sulphate (SDS) to protein ratios. Small volumes of an SDS stock solution were added to 20 μM cytochrome c solutions in 50 mM phosphate buffer, pH 6.8, to obtain the desire concentration of SDS. All experiments were performed at room temperature. The SDS to cytochrome c ratios are 0:1 (solid black line), 10:1 (dotted black line), 20:1 (dashed black line), 30:1 (solid grey line), 40:1 (dotted grey line) and 50:1 (dashed grey line)
Full SDS titration of wild-type, H26Q, H33N and H26Q/H33N cytochrome c
The titration of the four proteins was followed by visible spectroscopy and CD spectroscopy at pH 6.8. The dependence of the wavelength of the Soret band maximum on the SDS concentration is presented in Fig. 2. The Soret band maximum of wild-type cytochrome c remains constant at 408 nm at an SDS to cytochrome c ratio of up to approximately 100:1. This result is in agreement with a native low-spin state below the cmc [10, 20, 22]. The addition of SDS leads to the formation of a Soret band with a maximum at 402 nm, corresponding to an increase in the contribution of the high-spin form. At pH 6.8, further additions of SDS do not induce variations in the wavelength of the Soret band maximum; however, this maximum will be shown to be strongly pH dependent (vide infra).

Shift in the maximum absorbance of the Soret band as a function of increasing SDS concentration at pH 6.8 and room temperature. Symbols indicate the experimental data and the continuous lines were drawn for clarity. Small volumes of SDS were added to 20 μM cytochrome c solutions in 50 mM phosphate buffer, pH 6.8, to obtain the desired SDS concentration with wild-type (grey squares, grey line), H26Q (triangles, dashed black line), H33N (circles, solid black line) and H26Q/H33N (black squares, dotted black line) cytochrome c. cmc critical micelle concentration
The visible spectra of the double-mutant protein are also sensitive to SDS concentration. Below the cmc of SDS, a high-spin form with a Soret band maximum at 402 nm is observed. Further increases in the SDS concentration induce a blue shift to 398 nm, corresponding to another form of the high-spin state. The two obvious forms of the high-spin state correspond to pentacoordinated or hexacoordinated iron [17]. On the basis of previously reported variations in the Soret band maximum of insect haemoglobin [44, 45], the maxima at 402 and 398 nm were tentatively assigned to hexacoordinated (with a water molecule) and pentacoordinated iron, respectively. The main conclusion of the optical spectroscopy results is that the native state undergoes a direct transition from the native state to the high-spin state without any non-native low-spin intermediate. This highlights the role of the histidine in the formation of the non-native low-spin state.
The single-mutant proteins were constructed to evaluate the specific roles of His26 and His33. However, as shown in Fig. 2, both mutants behave like the wild-type protein below the cmc of SDS, with the formation of a non-native low-spin state. Clearly, this result demonstrates the ability of both His26 and His33 to replace the methionine after haem decoordination, leading to bishistidine species. Although no differences between the two histidines could be detected below the cmc of SDS, a surprising major difference was observed when the SDS concentration was increased. Both proteins undergo a spin-state transition towards the high-spin state. For H26Q human cytochrome c, this transition from the LSHH form to a high-spin form is characterized by a progressive shift of the Soret band, with a final Soret band maximum at 402 nm, very similar to the wild-type cytochrome c at pH 6.8. By contrast, for the H33N mutant, the final Soret band is strongly blue shifted, with a maximum at 398 nm, a value observed previously for the double mutant. These results suggest a specific role of His33 at pH 6.8; His33 may play a role in the transition between these two apparent high-spin states, which will be shown to be pH-sensitive (vide infra).
The low-field portions of the NMR spectra are shown in Fig. 3. Paramagnetic NMR is a powerful tool for discriminating between the various spin states of haemoproteins [46, 47]. Strongly downfield shifted signals at approximately 40–90 ppm, characteristic of haemoprotein in the high-spin form, are observed for both proteins. These peaks were assigned to the haem methyl protons of cytochrome c [17, 24]. The intensity of the signals reflects the level of the transition of cytochrome c to the high-spin state, which is dependent on the SDS concentration as previously observed by visible spectroscopy. For the wild-type protein (Fig. 3, spectra A), the signals are visible only above the cmc of SDS and reach a maximum at an SDS to cytochrome c ratio of 110:1. By contrast, the NMR spectra of the double mutant display two sets of paramagnetic signals (Fig. 3, spectra B). The first set, which is even visible at a low SDS to cytochrome c ratio, is linked to broad resonances that are slightly downfield shifted when compared with the second set present at higher micelle concentrations. Both sets are characteristic of two different high-spin species, which might be related to the presence or the absence of coordinated water or might be associated with structural modification due to the presence of SDS at micellar concentration.

1H NMR spectra of wild-type cytochrome c (A) and H26Q/H33N cytochrome c (B) at different SDS to cytochrome c ratios, recorded at 313 K. Small volumes of SDS-d 25 were added to a 0.1 mM cytochrome c solution in D2O (pH 6.8) to obtain the desired concentration
Influence of pH on the transitions
Because of the effect of pH on the first steps of the induction of apoptosis, which will be discussed later, careful pH titrations were also performed at different SDS concentrations (no SDS, SDS below the cmc and SDS above the cmc). The pH dependence of the Soret band maximum for the wild-type and the double-mutant proteins is shown in Fig. 4. Because the nature of the sixth ligand in the LSHX state is controversial and it may be a lysine [32–35], a large pH range was used. In the absence of micelles, the wild-type and double-mutant proteins exhibit similar behaviour throughout the pH titration. The Soret band maximum shifts from 409 nm at neutral pH to 406–407 nm under alkaline conditions, with pK of the classic alkaline transition occurring around pH 8.5 for both proteins. The sixth axial ligand at this pH was reported to be a lysine residue replacing the native Met80 [48].

Effect of pH on the wavelength of the Soret band maximum of cytochrome c; wild-type cytochrome c (A) and H26Q/H33N cytochrome c (B) at different SDS to cytochrome c ratios: 0:1 (solid line), 50:1 (dotted line) and 200:1 (dashed line). Small volumes of NaOH or HCl were added to 20 μM cytochrome c solutions in 50 mM phosphate buffer to obtain the desired pH. Spectra were recorded at room temperature
Below the cmc of SDS (with an SDS to cytochrome c ratio of 50:1), the LSHH form of the wild-type protein is observed in a pH range from 6.0 to 8.5 with a Soret band maximum at 408 nm. Further increases in pH result in a small blue shift of the Soret band maximum to approximately 406 nm, consistent with the lysine form. Below pH 6, some precipitation occurs, preventing fine analysis of the data. In the double mutant, there is no histidine as a potential ligand, and a Soret band maximum at approximately 406 nm is observed under basic conditions; it can be associated with a lysine as the sixth ligand. A decrease in pH induces a transition towards the formation of a high-spin species (Soret band maximum at 402 nm), which is complete at pH 6.8. This transition must be related to lysine protonation with an apparent pK much lower than that for free lysine characterizing better binding of lysine in the absence of competing histidine.
Above the cmc of SDS and under acidic conditions (pH 6), both proteins are in a high-spin state, with a Soret band maximum at approximately 398 nm. However, for the wild-type protein, the transition is not a continuous process, with an apparent inflexion point at approximately pH 6.8 and a Soret band maximum at approximately 402 nm, in good agreement with the data displayed in Fig. 3. A further decrease in pH induces a slow shift to 398 nm. When the pH is decreased, the double-mutant protein is progressively converted to the high-spin form characterized by a Soret band maximum at 398 nm. The apparent pK of 8.5 is similar to the previously reported pK for lysine observed for the alkaline transition of iso-1-ferricytochrome c [49]. This is consistent with a direct conversion of the lysine-bound form to the high-spin form. The Soret band maximum at 398 nm is different from that observed at a lower SDS to cytochrome c ratio. This change is associated with the larger structural variation induced at high SDS concentration. We have tentatively assigned the significance of this shift to the difference between the pentacoordinated and hexacoordinated (with a water molecule) high-spin forms.
The characterization of the different non-native cytochrome c forms at high SDS concentrations is improved by analysing the NMR data shown in Fig. 5. NMR spectra were acquired for the four proteins, and pH titrations were performed at a high SDS ratio (200:1). In Fig. 5, spectra A–D, the region from 15 to 80 ppm contains the paramagnetic cytochrome c haem methyl peaks of the four proteins studied. These peaks are highly sensitive to the spin state, between 50 and 80 ppm for the high-spin state and between 10 and 30 ppm for the low-spin state. Furthermore, among the various low-spin states, the chemical shifts are also sensitive to the nature of the sixth ligand. Under acidic conditions, at pH 5.6, the strongly downfield shifted signals, which were previously assigned to the haem methyl peaks of the high-spin form, are observed for the four proteins studied. However, the complete decrease in intensity of these signals, observed on increasing the pH, does not occur at the same pH. For the double-mutant cytochrome c, the decrease is correlated with the appearance of a peak at 26 ppm that displays full intensity at basic pH (Fig. 5, spectra D). This value corresponds to a low-spin species and is assigned to a haem methyl signal of the lysine-bound form. The low-field resonances of wild-type cytochrome c (Fig. 5, spectra A) and H26Q cytochrome c (Fig. 5, spectra B) disappear around pH 7.5, but the signal at 26 ppm is present at elevated pH. At intermediate pH, a new peak at 20.2 ppm is present, with a maximum intensity at pH 7.5, and is assigned to a haem methyl peak of the LSHH form. Comparing the integrations of the one high-spin haem methyl resonance with the peaks at 20.2 and 26 ppm reveals that each corresponding cytochrome c form is almost pure at a single pH. However, for these two proteins, the signal for the LSHH form, which is predominant at neutral pH, is also observed with the haem methyl signals of either the high-spin form or the lysine-bound form. The simultaneous presence of two different sets of haem signals is also indicative of the absence of fast exchange on the chemical shift timescale between the high-spin and LSHH forms as well as between the LSHK and LSHH forms. For H33N cytochrome c (Fig. 5, spectra C), the haem methyl signals are easily identified for the high-spin form under acidic conditions and the LSHK form, but the corresponding LSHH resonance is only weakly detectable. This broadening may be attributable to the highly dynamic behaviour of the cytochrome c mutant, indicating the weak ability of His26 to stabilize a well-defined low-spin state. This behaviour is confirmed by the results displayed in Fig. 5 (spectra E–H), which displays the CεH1 aromatic resonance of free histidine. After deuterium exchange of the amide protons, the CεH1 protons can be easily detected in the 8–9-ppm range. The signals are usually sharp; furthermore, 1H–13C HSQC spectra were acquired with a large coupling constant of 200 Hz to confirm the imidazole proton assignment in micellar solution (data not shown). Coordination of histidine to ferric iron prevents detection of any signals owing to severe broadening of the resonances induced by the paramagnetic environment. Consequently, detection of the aromatic histidine signals is only possible for free, unbound histidines. With the exception of the double-mutant protein, CεH1 aromatic resonances were observed for the three other proteins under strong acidic or strong basic conditions, corresponding to the free unbound histidines. The high similarity of the chemical shift values allows us to specifically assign each histidine at pH 5.6, with CεH1 at 8.7 and 8.8 for His26 and His33, respectively. Under basic conditions at pH 10.9, signals are observed at 7.88 and 7.81 for His26 and His33, respectively. These sharp peaks correspond to free protonated and unprotonated histidines, reflecting pure cytochrome c forms (high spin and LSHK) without any histidine coordination. During pH titration, two major effects are observed: (1) chemical shift variations indicating histidine pK values and (2) a decrease in the signal intensities, indicating histidine complexation. The decreases in the CεH1 aromatic intensities are directly related to the increase in the intensity of the peak at 20.2 ppm previously assigned to one haem methyl resonance of LSHH. During the titration of wild-type cytochrome c (Fig. 5, spectra E), both histidine proton intensities decrease, with a minimum around pH 7.2, corresponding to the apparent pK a obtained by chemical shift variation analysis of the full titration. However, the His33 aromatic proton suffers a more drastic intensity decrease than the corresponding His26 proton, suggesting that His33 has greater affinity than His26 for the haem iron. This difference is further confirmed by the study of the two mutants (Fig. 5, spectra F and G), in which the signal for the His33 proton nearly disappeared for H26Q cytochrome c, whereas the His26 proton signal is broadened and only partially decreased in intensity for the other mutant, H33N cytochrome c. These results are also consistent with the intensity of the 20.2-ppm resonance, which is very sharp for H26Q cytochrome c (Fig. 5, spectra B) but is broadened and cannot be detected for H33N cytochrome c (Fig. 5, spectra C). His33 is able to stabilize the LSHH form, leading to sharp haem methyl resonances. In contrast, in the H33N mutant spectra, a severe broadening of these resonances is observed, suggesting a strong dynamic behaviour of cytochrome c. Consequently, His26 does not appear to be as good a haem ligand as His33. This result is in agreement with the optical spectra displayed in Fig. 2, which show the difference between the two single-mutant proteins at SDS concentrations above the cmc. In the presence of SDS micelles at approximately neutral pH, histidine is the major ligand replacing the native methionine; the lysine adduct is observed only under more basic conditions.

1H NMR paramagnetic shifted region (left) and aromatic region (right) of wild-type (A, E), H26Q (B, F), H33N (C, G) and H26Q/H33N (D, H) human cytochrome c at different pH values and at a high SDS to cytochrome c ratio (200:1). The cytochrome c solutions (0.2 mM) were prepared in deuterated 50 mM phosphate buffer in the presence of 40 mM SDS-d 25. Small volumes of NaOD or DCl were added to obtain the desired pH. Spectra were recorded at 323 K
Discussion
Cytochrome c plays a major role in the early steps of apoptosis, spurring interest in the study of the interaction between cytochrome c and the inner mitochondrial membrane. In addition, newly developed drugs that act as ligands of the haem iron instead of the native methionine and prevent the peroxidase activity acquired by cardiolipin-bound cytochrome c are promising therapies [7, 50]. The membrane-bound form of cytochrome c has been studied in various model systems. The cytochrome c protein expands, with less-defined tertiary structure, upon the acquisition of a molten globule state [10, 21, 27, 51]. Decoordination of Met80 is associated with crevice opening, increasing both the volume and the solvent accessibility of the haem pocket [52]. In the literature, two different possible axial ligands have been proposed to replace the methionine after its decoordination, namely lysine [11, 34, 35] and histidine [10, 30, 31]. Because of differences in model systems and experimental conditions used previously and the high versatility of the coordination of cytochrome c, we have carefully studied the behaviour of cytochrome c under micellar conditions with special attention to the protein-to-micelle ratio and the effect of pH. To identify the putative different non-native cytochrome c low-spin species, cytochrome c mutants were designed and produced in the laboratory: H26Q, H33N and the double-mutant H26Q/H33N. We focused on histidine mutations because these two residues are good candidates for haem iron binding. SDS micelles have been shown to efficiently mimic the lipid interaction with cytochrome c by forming non-native low-spin and high-spin species. Furthermore, SDS is spectroscopically silent and limits the aggregation phenomena observed when using small unilamellar vesicles at physiologically relevant concentrations [53]. Finally, the influence of pH was carefully studied because variation of the pH has previously been implicated in the early stage of induction of apoptosis.
Behaviour of cytochrome c upon SDS titration at pH 6.8
The pH values of the mitochondrial intermembrane space, the cytosol and the mitochondrial matrix were estimated to be 6.9, 7.6 and 7.8, respectively [54, 55]. As cytochrome c is located in the mitochondrial intermembrane space, a pH of 6.8 was chosen in the first part of this study to approximate the physiological pH under non-apoptotic conditions. Significant differences between the wild-type and double-mutant proteins were observed upon addition of SDS at this pH. In the presence of SDS below the cmc, the wild-type and single-mutant proteins exhibit spectra characteristic of the low-spin state, in contrast to the double-mutant protein, which exhibits a characteristic high-spin-state spectrum. Both histidines are able to maintain cytochrome c in a low-spin state, definitively demonstrating the involvement of histidine in Met80 substitution in the membrane-mimic model.
Influence of pH on cytochrome c conformation
The forms of cytochrome c are sensitive to SDS concentration (below the cmc or above the cmc) and to pH. Thus, the influence of pH was carefully investigated at two different SDS concentrations over a large pH range, with particular care near the physiological pH. At SDS concentrations below the cmc, the LSHH form prevails over a large pH range from 6 to 8.5. However, below pH 6, some precipitation occurs, with the formation of large aggregates, as observed by differential interference contrast microscopy (data not shown). Such aggregation has been reported previously when cytochrome c is associated with anionic phospholipids under acidic conditions [56–58]. At SDS concentrations above the cmc, no aggregation occurs under acidic conditions. SDS micelles favour the wild-type cytochrome c in the high-spin form, with complete formation at pH 6 and lower. Formation of the LSHH form is complete when the pH is increased to 7.5. His33 is the major sixth axial ligand in this configuration rather than His26, most likely owing to reduced protein chain restriction. Interestingly, the balance between the high-spin form and the LSHH form is strongly dependent on the pH, which displays an amplitude of variation similar to that observed in the mitochondrial intermembrane space during apoptosis.
The transition towards the LSHK form begins at approximately pH 8 for H33N cytochrome c and the double mutant and at pH 8.5 for the wild-type and H26Q proteins. Remarkably, this transition is not SDS-dependent and is related to the deprotonation of the lysine residue. Generally, Lys73 and Lys79, which are close to Met80, are suggested to constitute the sixth axial ligand of the haem iron under basic conditions [49]. The controversy in the literature about the identity of the amino acid that replaces the native methionine is strongly associated with the experimental conditions. For many years, histidines have been proposed to be involved in iron complexation during the folding process [31, 59, 60] and membrane-mimetic interactions [10, 20, 22, 24]. Nevertheless, in the course of a study at pH 8.5 on chemically modified cytochrome c histidines, a non-native species was observed at an SDS to cytochrome c ratio of 70:1, which was obviously attributed to a lysine–histidine haem configuration (LSHK) [11]. This result is consistent with our data in this study, which show that the LSHK form was also encountered for the double-mutant protein, which has no histidine available to replace the methionine ligand as encountered when potential histidine ligands have been carbethoxylated. This former result has led to confusion in the literature, excluding the possibility of histidine binding and favouring lysine binding regardless of pH, as in Sinibaldi et al. [33, 34]. One of the aims of this study was to resolve the existing controversy in the literature by analysing the high cytochrome c polymorphism as a function of SDS concentration and pH. The use of histidine mutants was important as the differences between the LSHH and LSHK forms are very small, even with spectroscopic approaches. NMR experiments with the mutant proteins provided direct proof via a direct comparison study of the haem ligation state.
Model of the bishistidine form of cytochrome c (LSHH)
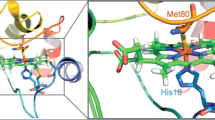
This study clearly demonstrated the presence of the LSHH form in SDS below or above the cmc. The LSHH form was modelled in silico (Fig. 6) with an interactive molecular simulation approach. This model is based on a well-defined native structure after steered modification of the 20-37 loop (see “Materials and methods”). This modification induced a haem tilt from the hydrophobic core of the protein to the histidine side, permitting the binding of His33. This effect corresponds to a large haem displacement away from Trp59, in agreement with the previously reported increase in tryptophan fluorescence [22]. This new haem c position is strongly stabilized by electrostatic interactions between the haem propionate groups and both Lys27 and Asn31. The proximity of the haem methyl groups to the junction of the N-terminal and C-terminal helices induces structural perturbations in the contact between these two helices. As this interaction is frequently proposed to play an essential role in the folding/unfolding of cytochrome c [59, 61], our LSHH model may represent a transient intermediate towards the fully flexible cytochrome c structure encountered in SDS, consistent with the loss of tertiary structure we observed by NMR spectroscopy and proposed in other studies [28, 62]. Further, the increase in haem solvent accessibility is also consistent with an increase in peroxidase activity.

Structural models of the native (LSHM, left) and the bishistidine (LSHH, right) forms of cytochrome c obtained by molecular dynamics relaxation. Molecular surface (top) and backbone (bottom) representations are coloured in blue (basic) or green (histidine) depending on the type of residue; the haem group is shown in red. The molecular hydrophobic potential is mapped on the molecular surface by a colour scale from white (polar) to black (hydrophobic) using the PLATINUM server [63]
Correlation with in vivo environment modifications occurring during apoptosis
During apoptosis, two major events in mitochondria affect the interaction of cytochrome c with the membrane. The first event is related to the pH variation that occurs before cytochrome c exits the mitochondria [64]. Proapoptotic stimuli such as Bax, stautosporine and UV irradiation induce mitochondrial matrix alkalinization (+0.5 pH units) and cytosol acidification (−0.5 pH units), followed by release of cytochrome c [54, 65]. This acidification of the mitochondrial intermembrane space induces F 0–F 1 pump inversion, which enhances acquisition of high spin by the membrane-bound form of cytochrome c. Moreover, the F 0–F 1 pump inversion leads to the hydrolysis of ATP to ADP. ATP has been shown to strongly inhibit the interaction of cytochrome c with the inner mitochondrial membrane [25, 66, 67]. Thus, a decrease in ATP concentration will increase the amount of cytochrome c tightly bound to the membrane. The second event is related to the cardiolipin migration that occurs during the first steps of the apoptotic process. Under physiological conditions, 20 % of the cardiolipin content is confined in the outer membrane, and this level increases to up to 40 % under apoptotic conditions. The inner membrane distribution is also sensitive to the condition, with a cardiolipin level inside the outer leaflet of 40 or 70 % for physiological and apoptotic conditions, respectively [2]. As the amount of cardiolipin increases near cytochrome c, the interaction between the two molecules increases. Binding of cytochrome c to phospholipid vesicles is predominantly electrostatic in nature through the interaction with the A site containing basic residues (Lys72, Lys73, Lys86, Lys87) or/and the L site (Lys22, Lys25, His26, Lys27, His33) [57, 68]. A hydrophobic acyl side chain could bind into the hydrophobic segment of cytochrome c at the C site [68] or close to Met80 [69]. Furthermore, the cytochrome c interaction is stronger when cardiolipin is partially protonated; the amount of this protonated form increases with acidic pH [70] and cardiolipin content [71]. At a high SDS to cytochrome c ratio, the transition between the LSHH form and the high-spin form occurs near the physiological pH, and even a small pH acidification leads to a large increase in the amount of the high-spin form, likely inducing higher peroxidase activity, generation of reactive oxygen species and escape of cytochrome c from the mitochondria to the cytosol.
Although the insertion of the acyl side chain may play a role in the peroxidase activity, the increase of the peroxidase-like activity of cytochrome c was reported to be associated with SDS concentrations higher than the cmc [72, 73].
Conclusion
We performed pH titrations on wild-type and mutant forms of human cytochrome c at different SDS concentrations. This strategy was used to mimic changing environmental conditions during apoptosis. This study unambiguously demonstrates the role of the histidine for methionine replacement in the non-native low-spin state. This study also demonstrates the predominance of the LSHH form compared with the proposed LSHK configuration, which predominates only at pH values greater than 8.5. This predominance is valid for both histidines at low SDS concentrations. On the basis of the NMR data, even at high SDS concentration, His33 is able to maintain the LSHH species throughout a large pH range; however, under slightly acidic conditions, a transition to the high-spin state is observed. In vivo, such mitochondrial intermembrane space acidification preceding exit of cytochrome c may drastically increase the amount of high-spin cytochrome c and consequently increase the peroxidase activity of cytochrome c. However, the LSHH form found at SDS concentrations below or above the cmc may act as a protective system that inhibits the acquisition of cytochrome c peroxidase activity when specific apoptotic conditions are not achieved.
References
Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Cell 86:147–157
Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, Greenberger JS, Pitt B, Shvedova AA, Borisenko G (2009) Free Radic Biol Med 46:1439–1453
Radi R, Turrens JF, Freeman BA (1991) Arch Biochem Biophys 288:118–125
Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG (2005) Nat Chem Biol 1:223–232
Yu T, Wang X, Purring-Koch C, Wei Y, McLendon GL (2001) J Biol Chem 276:13034–13038
Hanahan D, Weinberg RA (2011) Cell 144:646–674
Atkinson J, Kapralov AA, Yanamala N, Tyurina YY, Amoscato AA, Pearce L, Peterson J, Huang Z, Jiang J, Samhan-Arias AK, Maeda A, Feng W, Wasserloos K, Belikova NA, Tyurin VA, Wang H, Fletcher J, Wang Y, Vlasova II, Klein-Seetharaman J, Stoyanovsky DA, Bayir H, Pitt BR, Epperly MW, Greenberger JS, Kagan VE (2011) Nat Commun 2:497
de Jongh HH, Ritsema T, Killian JA (1995) FEBS Lett 360:255–260
Zucchi MR, Nascimento OR, Faljoni_Alario A, Prieto T, Nantes IL (2003) Biochem J 370:671–678
Oellerich S, Wackerbarth H, Hildebrandt P (2002) J Phys Chem B 106:6566–6580
Mugnol KC, Ando RA, Nagayasu RY, Faljoni-Alario A, Brochsztain S, Santos PS, Nascimento OR, Nantes IL (2008) Biophys J 94:4066–4077
Muga A, Mantsch HH, Surewicz WK (1991) Biochemistry 30:7219–7224
Spooner PJ, Watts A (1991) Biochemistry 30:3880–3885
Heimburg T, Hildebrandt P, Marsh D (1991) Biochemistry 30:9084–9089
Pinheiro TJ, Watts A (1994) Biochemistry 33:2451–2458
Rytomaa M, Mustonen P, Kinnunen PK (1992) J Biol Chem 267:22243–22248
Oellerich S, Wackerbarth H, Hildebrandt P (2003) Eur Biophys J 32:599–613
Keilin D, Hartree EF (1940) Nature 145:934
Takeda K, Takahashi K, Batra PP (1985) Arch Biochem Biophys 236:411–417
Yoshimura T (1988) Arch Biochem Biophys 264:450–461
de Jongh HH, Killian JA, De Kruijff B (1992) Biochemistry 31:1636–1643
Das TK, Mazumdar S, Mitra S (1998) Eur J Biochem 254:662–670
Gebicka L, Gebicki JL (1999) J Protein Chem 18:165–172
Chevance S, Le_Rumeur E, de_Certaines JD, Simonneaux G, Bondon A (2003) Biochemistry 42:15342–15351
Ahluwalia U, Nayeem SM, Deep S (2011) Eur Biophys J 40:259–271
Vladimirov YA, Proskurnina EV, Izmailov DY, Novikov AA, Brusnichkin AV, Osipov AN, Kagan VE (2006) Biochemistry (Mosc) 71:989–997
Bertini I, Turano P, Vasos PR, Bondon A, Chevance S, Simonneaux G (2004) J Mol Biol 336:489–496
Hanske J, Toffey JR, Morenz AM, Bonilla AJ, Schiavoni KH, Pletneva EV (2012) Proc Natl Acad Sci USA 109:125–130
Domanov YA, Molotkovsky JG, Gorbenko GP (2005) Biochim Biophys Acta 1716:49–58
Colon W, Wakem LP, Sherman F, Roder H (1997) Biochemistry 36:12535–12541
Muthukrishnan K, Nall BT (1991) Biochemistry 30:4706–4710
Nantes IL, Mugnol KC (2008) J Liposome Res 18:175–194
Sinibaldi F, Droghetti E, Polticelli F, Piro MC, Di Pierro D, Ferri T, Smulevich G, Santucci R (2011) J Inorg Biochem 105:1365–1372
Sinibaldi F, Howes BD, Piro MC, Polticelli F, Bombelli C, Ferri T, Coletta M, Smulevich G, Santucci R (2010) J Biol Inorg Chem 15:689–700
Bradley JM, Silkstone G, Wilson MT, Cheesman MR, Butt JN (2011) J Am Chem Soc 133:19676–19679
Jeng WY, Chen CY, Chang HC, Chuang WJ (2002) J Bioenerg Biomembr 34:423–431
Wegerich F, Turano P, Allegrozzi M, Mohwald H, Lisdat F (2009) Anal Chem 81:2976–2984
Gourion-Arsiquaud S, Chevance S, Bouyer P, Garnier L, Montillet J-L, Bondon A, Berthomieu C (2005) Biochemistry 44:8652–8663
Delalande O, Ferey N, Grasseau G, Baaden M (2009) J Comput Chem 30:2375–2387
Krieger E, Darden T, Nabuurs SB, Finkelstein A, Vriend G (2004) Proteins 57:678–683
Harbury HA, Cronin JR, Fanger MW, Hettinger TP, Murphy AJ, Myer YP, Vinogradov SN (1965) Proc Natl Acad Sci USA 54:1658–1664
Smith DW, Williams RJ (1970) Struct Bonding 7:1–45
Myer YP (1968) J Biol Chem 243:2115–2122
Kamimura S, Matsuoka A, Imai K, Shikama K (2003) Eur J Biochem 270:1424–1433
MarmoMoreira L, LimaPoli A, Costa-Filho AJ, Imasato H (2006) Biophys Chem 124:62–72
Bertini I, Turano P, Vila AJ (1993) Chem Rev 93:2833–2932
La Mar GN, de Ropp JS (1993) In: Berliner LJ, Reuben J (eds) NMR of paramagnetic molecules. Plenum, New York, pp 1–78
Assfalg M, Bertini I, Dolfi A, Turano P, Mauk AG, Rosell FI, Gray HB (2003) J Am Chem Soc 125:2913–2922
Dopner S, Hildebrandt P, Rosell FI, Mauk AG (1998) J Am Chem Soc 120:11246–11255
Kagan VE, Wipf P, Stoyanovsky D, Greenberger JS, Borisenko G, Belikova NA, Yanamala N, SamhanArias AK, Tungekar MA, Jiang J, Tyurina YY, Ji J, Klein-Seetharaman J, Pitt BR, Shvedova AA, Bayir H (2009) Adv Drug Deliv Rev 61:1375–1385
Pinheiro TJT, Elöve GA, Watts A, Roder H (1997) Biochemistry 36:13122–13132
Sanghera N, Pinheiro TJT (2000) Protein Sci 9:1194–1202
Bernad S, Oellerich S, Soulimane T, Noinville S, Baron MH, Paternostre M, Lecomte S (2004) Biophys J 86:3863–3872
Matsuyama S, Llopis J, Deveraux QL, Tsien RY, Reed JC (2000) Nat Cell Biol 2:318–325
Porcelli AM, Ghelli A, Zanna C, Pinton P, Rizzuto R, Rugolo M (2005) Biochem Biophys Res Commun 326:799–804
Kawai C, Pessoto FS, Rodrigues T, Mugnol KC, Tortora V, Castro L, Milicchio VA, Tersariol IL, Di Mascio P, Radi R, Carmona-Ribeiro AM, Nantes IL (2009) Biochemistry 48:8335–8342
Kawai C, Prado FM, Nunes GL, Di Mascio P, Carmona-Ribeiro AM, Nantes IL (2005) J Biol Chem 280:34709–34717
Zhao H, Tuominen EK, Kinnunen PK (2004) Biochemistry 43:10302–10307
Colon W, Roder H (1996) Nat Struct Biol 3:1019–1025
Latypov RF, Cheng H, Roder NA, Zhang J, Roder H (2006) J Mol Biol 357:1009–1025
Konermann L, Pan J, Liu YH (2010) Chem Soc Rev 40:1224–1234
Hirota S, Hattori Y, Nagao S, Taketa M, Komori H, Kamikubo H, Wang Z, Takahashi I, Negi S, Sugiura Y, Kataoka M, Higuchi Y (2010) Proc Natl Acad Sci USA 107:12854–12859
Pyrkov TV, Chugunov AO, Krylov NA, Nolde DE, Efremov RG (2009) Bioinformatics 25:1201–1202
Lagadic-Gossmann D, Huc L, Lecureur V (2004) Cell Death Differ 11:953–961
Matsuyama S, Reed JC (2000) Cell Death Differ 7:1155–1165
Tuominen EK, Zhu K, Wallace CJ, Clark-Lewis I, Craig DD, Rytomaa M, Kinnunen PK (2001) J Biol Chem 276:19356–19362
Patriarca A, Eliseo T, Sinibaldi F, Piro MC, Melis R, Paci M, Cicero DO, Polticelli F, Santucci R, Fiorucci L (2009) Biochemistry 48:3279–3287
Rytömaa M, Kinnunen PKJ (1994) J Biol Chem 269:1770–1774
Kalanxhi E, Wallace CJ (2007) Biochem J 407:179–187
Gorbenko GP, Molotkovsky JG, Kinnunen PK (2006) Biophys J 90:4093–4103
Trusova VM, Gorbenko GP, Molotkovsky JG, Kinnunen PK (2010) Biophys J 99:1754–1763
Diederix REM, Busson S, Ubbink M, Canters GW (2004) J Mol Cat B 27:75–82
Gebicka L, Didik J (2005) Acta Biochim Pol 52:551–555
Acknowledgments
The human cytochrome c plasmid and purified wild-type human cytochrome c were generously provided by W.Y. Jeng. Escherichia coli cells were a gift from P. Turano. We also thank S. Pastezeur for gene sequencing, J. Jardin for mass spectrometry measurements and G. Gambarota for careful reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
The supplementary material includes full details of the preparation of mutated proteins and two figures. Figure S1: identification of the different iron spin state limit forms of the wild-type human ferricytochrome c at 20 μM in 50 mM phosphate buffer, pH 6.8. Figure S2: various forms of cytochrome c.
Rights and permissions
About this article
Cite this article
Simon, M., Metzinger-Le Meuth, V., Chevance, S. et al. Versatility of non-native forms of human cytochrome c: pH and micellar concentration dependence. J Biol Inorg Chem 18, 27–38 (2013). https://doi.org/10.1007/s00775-012-0946-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-012-0946-4