
Abstract
Hephaestin is a multicopper ferroxidase expressed mainly in the mammalian small intestine. The ferroxidase activity of hephaestin is thought to play an important role during iron export from intestinal enterocytes and the subsequent iron loading of the blood protein transferrin, which delivers iron to the tissues. Structurally, the ectodomain of hephaestin is predicted to resemble ceruloplasmin, the soluble ferroxidase of blood. In this study, the human hephaestin ectodomain was expressed in baby hamster kidney cells and purified to electrophoretic homogeneity. Ion exchange chromatography of purified recombinant human hephaestin (rhHp) resulted in the isolation of hephaestin fractions with distinct catalytic and spectroscopic properties. The fraction of rhHp with the highest enzymatic activity also showed an enhanced molar absorptivity at 600 nm, characteristic of type 1 copper sites. Kinetic analysis revealed that rhHp possesses both high-affinity and low-affinity binding sites for ferrous iron. To investigate the role of particular residues in iron specificity of hephaestin, mutations of putative iron ligands were introduced into rhHp using site-directed mutagenesis. Kinetic analysis of ferroxidation rates of wild-type rhHp and mutants demonstrated the important roles of hephaestin residues E960 and H965 in the observed ferroxidase activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mammalian hephaestins are a group of multicopper ferroxidases expressed predominantly in the small intestine [1, 2]. Localized on the basolateral surface of enterocytes, hephaestin oxidizes Fe(II) exported by the basolateral ferrous transporter ferroportin to Fe(III) (for a review see [3]). Through this oxidation step, iron is made bioavailable for binding to transferrin, the major iron transporter in blood that specifically binds ferric ions [4]. The importance of this hephaestin ferroxidase function is illustrated by the sla mouse phenotype—such mice have an in-frame deletion in the heph gene resulting in a truncated form of hephaestin that shows significantly decreased ferroxidase activity compared with the wild-type protein [5]. The subsequent impaired iron export from enterocytes into blood gives a deficiency of iron in the tissues (especially hematopoietic cells, which are the major consumers of iron). Thus, mice carrying the sla mutation develop a microcytic hypochromic anemia with iron accumulation in the intestinal epithelium [6]. Although hephaestin is mainly expressed in the intestine, this protein has recently been found in heart, brain, and pancreas [7, 8]. In these tissues, the hephaestin ferroxidase activity may help to minimize the Fe2+ toxicity resulting from the formation of free radicals through the Haber–Weiss-Fenton series of reactions [9].
The predicted amino acid sequence of human hephaestin is 50 % identical and 68 % similar to the sequence of human ceruloplasmin, the major multicopper ferroxidase of blood [10]. Although both proteins contain six cupredoxin domains, hephaestin also has a predicted transmembrane domain at the C terminus [10]. On the basis of the known crystal structure of ceruloplasmin, comparative structural modeling of the hephaestin ectodomain revealed that with the exception of a type 1 copper ligand in domain 2, all residues involved in copper binding in ceruloplasmin are conserved in hephaestin [10]. Both paralogs contain ligands for three types of copper sites. The type 1 (“blue”) copper site serves as an acceptor of electrons from the substrate; as a result of a Cys(S−) to Cu2+ charge transfer, the presence of copper in the type 1 site is associated with an intense absorption at 600 nm (ε 600 nm = 4,500–6,000 M−1 cm−1) [11]. The type 2 copper site and the binuclear type 3 copper site form a trinuclear cluster and are the site of dioxygen binding and reduction. Through electron transfer from the type 1 copper sites to the trinuclear copper cluster, one-electron oxidation of four substrate equivalents is coupled with the four-electron reduction of O2 to H2O. Similar to ceruloplasmin, hephaestin is predicted to contain six copper atoms per molecule: three atoms that are present in the type 1 copper sites in domains 2, 4, and 6, and three atoms that are present in a trinuclear cluster on the interface of domains 1 and 6 [10]. In hephaestin, methionine, cysteine, and two histidine residues are putative copper ligands for all three type 1 copper atoms (Table 1). Although the same arrangement of type 1 copper ligands is observed in ceruloplasmin domains 4 and 6 [12], ceruloplasmin domain 2 contains a leucine residue instead of a methionine (Table 1), resulting in the unusually high redox potential of this site [13].
On the basis of the results of crystal soaking experiments [12], ceruloplasmin contains two ferrous binding sites located in the vicinity of the type 1 copper atoms in domains 4 and 6. Two glutamic acid residues, one aspartic acid residue, and one histidine residue are iron ligands of both sites [12] (Table 2). Crystal soaking experiments did not reveal ferrous iron binding in domain 2 of ceruloplasmin, which contains E236, E633, N323, and Y241 in the corresponding positions. Compared with ceruloplasmin, hephaestin contains conserved ligands in the Fe(II) binding site of domain 6 (E960, H965, D1050, E300), whereas in domains 2 and 4, the equivalent residues are E264, H269, S351, and E652 and D616, H621, S703, and D996, respectively (Table 2). In addition to Fe(II), both hephaestin and ceruloplasmin can oxidize organic substrates such as p-phenylenediamine (pPD) [14, 15]. As shown by crystal soaking experiments, ceruloplasmin binds pPD at the base of domain 4, which is remote from the type 1 copper site in this domain [16].
Although hephaestin has never been isolated from human tissue, we have previously expressed the recombinant hephaestin ectodomain in baby hamster kidney (BHK) cells [17]. Recombinant human hephaestin (rhHp) produced in this expression system showed lower ferroxidase activity compared with other multicopper ferroxidases [17]. As shown by inductively coupled plasma mass spectrometry, the copper content was 3.13 copper atoms per molecule of rhHp instead of the predicted six atoms per molecule of rhHp [17]. The spectroscopic properties of rhHp (ε 607 nm = 2,010 M−1) were consistent with an average of approximately 0.5 type 1 copper sites per molecule of hephaestin [17]. All these observations suggested the incomplete copper loading of rhHp in this expression system. Furthermore, the use of the yeast Pichia pastoris as an expression system did not result in production of rhHp with the predicted copper content. Instead, this yeast-derived rhHp contained only 4.2 copper atoms per molecule [14].
In the current study, we used ion exchange chromatography (IEC) to fractionate hephaestin into several fractions of rhHp that displayed different catalytic and spectroscopic properties. By using site-directed mutagenesis, we introduced mutations of the putative Fe(II)-binding residues in rhHp. The kinetics of the ferroxidase and p-phenylenediamine oxidase activities of wild-type rhHp and mutants was investigated. These studies provide the first experimental evidence for the functional role of individual amino acid residues in iron oxidation by human hephaestin.
Materials and methods
Materials
Oligonucleotide synthesis was performed by the Nucleic Acid Protein Service (NAPS) at the University of British Columbia and Integrated DNA Technologies (San Diego, CA, USA). Enzymes for DNA manipulation were from New England Biolabs (Beverly, MA, USA). All other reagents were purchased from Sigma-Aldrich (Oakville, ON, Canada) unless otherwise noted.
DNA manipulation
A modified hephaestin complementary DNA [17] that had been incorporated into the pBSSK− vector (Stratagene, La Jolla, CA, USA) via NotI restriction sites was used as an initial construct for DNA manipulation. Modifications previously introduced to the hephaestin complementary DNA include replacement of the native signal peptide by the transferrin signal sequence and substitution of the predicted transmembrane domain with a 1D4 epitope at the C terminus [17]. For the current studies, the transferrin signal sequence was replaced with the human ceruloplasmin signal sequence using a QuikChange mutagenesis kit (Stratagene, La Jolla, CA, USA) and the primers CpSignal-F and CpSignal-R (Table S1). The DNA fragment encoding the hephaestin ectodomain (with the ceruloplasmin signal peptide and 1D4 epitope at the C terminus) was subcloned into the pNUT vector [18] using NotI restriction sites. The resulting construct was used for expression of rhHp and as a template for subsequent site-directed mutagenesis.
Mutations of putative iron ligands in the hephaestin sequence were introduced by using the megaprimer technique [19] and primers E264A/H269A-R, E616A/H621A-F, E960A/H965A-F, pNUT5′, and pNUT3′ (see Table S1). For propagating DNA, Escherichia coli Mach1 cells were grown in Luria–Bertani medium supplemented with ampicillin (100 mg L−1). Plasmid DNA and PCR products were purified by using plasmid DNA miniprep kits and gel extraction kits (Epoch Biolabs, Missouri City, TX, USA). Automated DNA sequence analysis using a BigDye Terminator kit (Stratagene, La Jolla, CA, USA) and an ABI 3700 DNA sequencer (Applied Biosystems, Streetsville, ON, Canada) was used to verify the nucleotide sequences of all constructs on both strands of DNA.
Expression and purification of rhHp and mutants
BHK cells were grown to confluence in six-well tissue culture plates in Dulbecco’s modified Eagle’s medium–Ham F12 nutrient mixture (DMEM-F12) containing 5 % newborn calf serum (Invitrogen, Burlington, ON, Canada) in a humidified 5 % CO2 atmosphere. Transfection of BHK cells with recombinant DNA was performed using the FuGene 6 transfection reagent according to the manufacturer’s instructions (Roche, Indianapolis, IN, USA). One day after transfection, methotrexate was added to the medium to a final concentration of 200 mg L−1. After 14 days of growth on selective medium, colonies of methotrexate-resistant cells were expanded into flasks for further transfer into expanded surface roller bottles (1,700 cm2; Fisher Scientific, Ottawa, ON, Canada). When cells in the roller bottle reached confluence, the DMEM-F12–newborn calf serum–methotrexate medium was replaced with DMEM-F12 containing 2 % Ultroser G (BioSepra, Marlborough, MA, USA) and 10 μM CuSO4. Subsequent batches contained DMEM-F12, 1 % Ultroser G, and 10 μM CuSO4 (200 mL per roller bottle). The culture medium containing Ultroser G was collected every 2 days for subsequent recombinant protein purification.
Monoclonal antibodies to the 1D4 epitope [20] were coupled to Sepharose 2B by using the CNBr activation method described by Cuatrecasas [21]. The culture medium was clarified by centrifugation and passed through the immunoaffinity column, which had been equilibrated with 20 mM tris(hydroxymethyl)aminomethane (Tris–HCl), 150 mM NaCl, pH 7.4 (storage buffer). The column was washed with ten bed volumes of the storage buffer. Elution was performed by repetitive application of 0.1 mg mL−1 1D4 peptide (N-Ac-TETSQVAPA; purchased from BioBasics, Markham, ON, Canada) diluted in storage buffer. The elution fractions were concentrated using Amicon Ultra-15 centrifugal filter units with a 30-kDa cutoff (Millipore, Billerica, MA, USA). The purity of the eluted protein in the fractions was confirmed by 10 % sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by staining with 0.1 % Coomassie blue.
IEC of rhHp
Immunoaffinity-purified rhHp (1 mg) in low-salt buffer (5 mM Tris–HCl 15 mM NaCl, pH 7.4) was applied onto the column with diethylaminoethyl (DEAE)-Sepharose (bed volume 1 mL). The column was then washed with ten bed volumes of the low-salt buffer. Elution was performed by stepwise increase of the salt concentration. Each elution step involved application of 1.5 ml of 5 mM Tris–HCl, pH 7.4 buffer containing 150, 200, and 250 mM NaCl. Prior to analysis, the eluted fractions were buffer-exchanged against the storage buffer (20 mM Tris–HCl 150 mM NaCl, pH 7.4) using Amicon Ultra-15 centrifugal filter units with a 30-kDa cut off.
Analytical methods
For N-terminal sequence analysis, 3 μg of rhHp was electrophoresed on 10 % sodium dodecyl sulfate polyacrylamide gel and then transferred to a poly(vinylidene difluoride) membrane (Bio-Rad, Mississauga, ON, Canada) in N-cyclohexyl-3-aminopropanesulfonic acid buffer according to the manufacturer’s instructions. The membrane was stained with 0.1 % Coomassie blue. The band representing rhHp was excised and analyzed with an ABI 492 Procise cLC sequencer at the Hospital for Sick Children in Toronto (http://www.sickkids.ca). UV–vis absorbance spectra of rhHp samples in 20 mM Tris–HCl, 150 mM NaCl buffer, pH 7.4 were recorded with a Varian Cary 4000 spectrophotometer (25 °C). The concentration of rhHp was determined using the experimental molar absorption coefficient for the absorbance at 280 nm (215,474 M−1 cm−1) [17]. Inductively coupled plasma mass spectrometry using a dynamic reaction cell was performed by Applied Speciation and Consulting (Bothell, WA, USA; http://www.appliedspeciation.com) according to its established procedures.
Activity assays
All activity assays were performed at room temperature in 96-well plates (Corning, Corning, NY, USA). As an autooxidation control, equivalent amounts of the buffer were used instead of the protein solution. Kinetic parameters were calculated using gnuplot (http://www.gnuplot.info).
The ferroxidase activity assay was performed in 75 mM sodium acetate buffer, pH 5 with a total reaction volume of 200 μL. A defined amount of ferrous ammonium sulfate was added to each reaction mixture. After incubation with either protein solution or elution buffer, reactions were quenched with 50 μL of 4 mM ferrozine, an Fe(II)-specific chelator. The ferrozine–Fe(II) complex has a strong absorbance at 562 nm (ε 562 nm = 27.9 mM−1 cm−1 [22]), which was measured using a microplate reader. The concentration of Fe(II) before and after incubation was determined using a calibration curve that was linear for the Fe(II) concentration range of 2–100 μM. For determination of kinetic parameters, the initial kinetic data were plotted in Eadie–Hofstee coordinates.
Oxidation of p-phenylenediamine dihydrochloride was assayed in 250 μL of 80 mM sodium acetate buffer containing 80 μM EDTA, pH 5. EDTA was added to the reaction to prevent iron-mediated oxidation of pPD [14, 23]. The rates of pPD oxidation were calculated using absorption coefficients for Bandrowski’s base (ε 535 nm = 1,910 M−1 cm−1), which is the product of pPD oxidation [24]. Because production of a single molecule of Bandrowski’s base involves oxidation of three pPD molecules [16], the absorbance-based oxidation rates were multiplied by 3, and the resultant catalytic rates were expressed as the number of pPD molecules oxidized by one molecule of enzyme per minute. For determination of K m and k cat, pPD oxidation rates were measured at a minimum of eight substrate concentrations ranging from 0.05 to 7.5 mM. The initial reaction rates were directly fitted to the Michaelis–Menten equation using nonlinear regression.
Results
Expression and initial characterization of rhHp
Hephaestin ectodomain with ceruloplasmin signal peptide was expressed in BHK cells. The 1D4 epitope introduced at the C terminus of hephaestin allowed the purification of rhHp with a single immunoaffinity chromatography step. N-terminal sequence analysis of rhHp revealed the following sequence: (A/S/G)-T-R-V-Y-Y. Despite the ambiguous identity of the first amino acid, the remainder of the sequence (along with the first residue when alanine is included) corresponds to the amino acid sequence of human hephaestin. In contrast to a previous report, where hephaestin was reported to contain additional amino acids from the transferrin signal peptide [17], the rhHp used in this study had an authentic human hephaestin N-terminal sequence.
When the ferroxidase and p-phenylenediamine oxidase activities of rhHp were analyzed, it appeared that samples of hephaestin purified from different batches of medium had significant variability in specific activity. A correlation between the p-phenylenediamine oxidase and ferroxidase activities of rhHp samples was observed (Fig. 1, circles in the lower-left quadrant), suggesting that both types of enzymatic activity originate from the same protein species. Despite the variability in specific activity, the K m values for pPD and Fe2+ were consistent throughout the samples (Table S2). These results suggest that there may be a variation in the amount of enzymatically active hephaestin in different samples.

Correlation between the specific p-phenylenediamine oxidase (pPD-oxidase) and ferrioxidase activities of different recombinant human hephaestin (rhHp) samples. Both p-phenylenediamine oxidase and ferroxidase activities are expressed as units of activity (U). p-Phenylenediamine oxidase activity was assayed in 0.8 mM sodium acetate, 80 μM EDTA, 1.5 mM p-phenylenediamine (pPD), pH 5, total reaction volume 250 μL; 1 U is defined as 1 nmol of pPD per liter oxidized per minute by 1 μg of rhHp. Ferroxidase activity was assayed in 0.75 mM sodium acetate, 10 μM ferrous ammonium sulfate, pH 5, total reaction volume 200 μL; 1 U is defined as 1 nmol of Fe2+ per liter oxidized per minute by 1 μg of rhHp. All activity assays were performed in triplicate, with the mean values shown in the plot. Circles samples of unfractionated rhHp, Diamonds rhHp samples eluted during ion exchange chromatography at 150 mM NaCl
Fractionation of purified hephaestin by IEC
In previous studies, IEC was used to separate apoceruloplasmin and holoceruloplasmin [25]. During elution with increasing salt concentration, holoceruloplasmin was eluted first and the apoceruloplasmin was recovered at a higher ionic strength [25]. We applied a similar procedure to further fractionate rhHp purified by the immunoaffinity chromatography step. Purified hephaestin was loaded onto DEAE-Sepharose in a low-salt buffer. After unbound protein had been removed with the same low-salt buffer, a stepwise salt gradient was applied. The fractions of hephaestin eluted at different salt concentrations were buffer-exchanged against storage buffer and concentrated. Sodium dodecyl sulfate polyacrylamide gel electrophoresis confirmed that only full-size hephaestin with apparent molecular mass of 130,000 Da was eluted (Fig. 2).

Fractionation of rhHp by chromatography on diethylaminoethyl (DEAE)-Sepharose. 1 rhHp prior to chromatography, 2 rhHp fraction eluted at 150 mM NaCl, 3 rhHp fraction eluted at 200 mM NaCl, 4 rhHp fraction eluted at 250 mM NaCl. a specific p-phenylenediamine oxidase activity. Error bars represent the standard error of the mean. b sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie staining; each lane contains 1 μg of rhHp. On the basis of SDS-PAGE, rhHp had an apparent molecular mass of 130 kDa, whereas the molecular mass predictedfor rhHp is 119 kDa. c copper content of rhHp expressed as the number of copper atoms per molecule. Copper content of protein solutions was determined by inductively coupled plasma mass spectrometry with dialysis buffer used as a control. Errors represent the range of duplicate measurements
Subsequent analysis of the specific p-phenylenediamine oxidase activity showed that the fraction of hephaestin eluted at 150 mM NaCl had higher activity compared with the hephaestin sample prior to IEC as well as the other fractions eluted at higher salt concentrations (Fig. 2). We then performed a kinetic analysis of this highly enzymatically active fraction. Initial ferroxidation rates were measured at iron concentrations ranging from 2 to 10 μM. Kinetic analysis revealed the following parameters for IEC-purified rhHp: for Fe(II), K m = 4.0 ± 1.5 μM and k cat = 18 ± 3 min−1; for pPD, K m = 1.5 ± 0.2 mM and k cat = 29 ± 1 min−1. This DEAE-Sepharose fractionation of rhHp was performed in two independent experiments, and in both cases the specific activity of the rhHp fraction eluted at 150 mM (Fig. 1, diamonds in the upper-right quadrant) was significantly higher than the specific activities of unfractionated rhHp samples (Fig. 1, circles in the lower-left quadrant).
We also measured the UV–vis absorption spectra of hephaestin fractions eluted from the DEAE-Sepharose column at different salt concentrations. All fractions showed a distinguishable peak at 600 nm characteristic of a type 1 copper site, with the hephaestin fraction eluted at 150 mM NaCl having the highest absorption coefficient of ε 600 nm = 5,500 M−1 cm−1 (Fig. 3). Although differing in enzymatic activity and spectroscopic properties, the hephaestin fractions did not show significant differences in copper content (Fig. 2). This was unexpected but could be the result of adventitious (nonspecific) binding of nonfunctional copper to the rhHp species.

Spectroscopic properties of rhHp fractions produced by chromatography on DEAE-Sepharose. Absorption spectra of protein solutions were determined against a blank comprising dialysis buffer (20 mM Tris–HCl, 150 mM NaCl, pH 7.4). Absorbance is given as the molar absorptivity calculated from spectra normalized for protein concentration. As addition of oxidant to rhHp did not result in an increase of absorbance at 600 nm (data not shown), hephaestin produced in the baby hamster kidney cell expression system is expected to contain only fully oxidized type 1 copper site(s). Straight lines rhHp fraction eluted at 150 mM NaCl, dashed lines rhHp fraction eluted at 200 mM NaCl, dotted lines rhHp fraction eluted at 250 mM NaCl
Kinetic analysis of hephaestin-catalyzed ferroxidation
In previous studies, the ferroxidase activity of hephaestin was analyzed only at low substrate concentrations [14, 17]. Under these conditions, hephaestin exhibited Michaelis–Menten-type kinetics for the oxidation of Fe(II). In the current study, we determined the hephaestin-catalyzed ferroxidation rates at a wide range of iron concentrations. This revealed non-Michaelis–Menten kinetics of hephaestin ferroxidase activity that is especially noticeable when the kinetic data are plotted in Eadie–Hofstee coordinates (Fig. 4). In the case of rhHp, the Eadie–Hofstee plot of the initial kinetic data corresponds to a biphasic curve instead of the single line predicted by the Michaelis–Menten equation. A two-component Eadie–Hofstee plot suggests the presence of two types of iron-binding sites with different affinities for Fe(II). Thus, the resultant kinetic parameters of rhHp ferroxidase activity are K m = 3.5 ± 0.2 μM and V max = 67 ± 2 nM min−1 for the high-affinity binding site(s) and K m = 107 ± 13 μM and V max = 286 ± 16 nM min−1 for the low-affinity binding site(s).

Ferroxidase activity of recombinant hephaestin. a Velocity versus substrate plot for rhHp. Initial ferroxidation rates were determined using a discontinuous ferrozine assay. Reaction mixtures containing 0.02 μM rhHp (95 U of p-phenylenediamine oxidase activity, 173 U of ferroxidase activity) were incubated with various concentrations of ferrous iron. After incubation for 42 min, reactions were quenched with excess ferrozine. For iron concentrations greater than 100 μM, an aliquot of reaction mixture was taken and diluted tenfold prior to quenching with ferrozine; these dilutions brought the absorbance values into the detectable range of the microplate reader. b Kinetic data from a plotted in Eadie–Hofstee coordinates. The Eadie–Hofstee plot was used to determine kinetic parameters for high-affinity and low-affinity binding sites. Dashed lines kinetic curve (a) and line on Eadie–Hofstee plot (b) modeled for a high-affinity binding site, dot-dashed lines kinetic curve (a) and line on Eadie–Hofstee plot (b) modeled for a low-affinity ferrous binding site. All reactions were performed in sextuplicate. Error bars represent the standard error of the mean
Site-directed mutagenesis of putative iron ligands in rhHp
Amino acid sequence homology with ceruloplasmin predicted that hephaestin has three putative iron-binding sites in domains 2, 4, and 6 [10]. To study the role of each binding site in the ferroxidase activity of hephaestin, we produced the following mutants: IB (E264A/H269A; D616A/H621A; E960A/H965A), IB6 (E264A/H269A; D616A/H621A), IB4 (E264A/H269A; E960A/H965A), and IB2 (D616A/H621A; E960A/H965A). All three iron-binding sites are mutated in the IB mutant, whereas mutants IB6, IB4, and IB2 have a single unaffected binding site in domains 6, 4, and 2, respectively. By mutating residues in two iron-binding sites at once (mutants IB6, IB4 and IB2), we studied the impact of the unaffected site on ferroxidase activity of rhHp.
Similar to the wild-type rhHp characterized in this study, all hephaestin mutants were expressed in BHK cells and purified by immunoaffinity chromatography. To ensure that the effects of the mutations on ferroxidase activity of hephaestin were not the result of protein misfolding, we performed kinetic analyses of the pPD-oxidation rates for rhHp and mutants. All mutants retained gross p-phenylenediamine oxidase activity but showed lower K m values compared with K m for pPD of rhHp (Table 3, columns 2 and 3), suggesting that the gross protein conformations were preserved and the resulting differences in ferroxidase activity were reflective of the mutations that had been introduced into the predicted iron-binding sites. Notably, despite the variability of the k cat values in different samples of the same rhHp species, the K m values and the ratios between the pPD- and Fe(II)-oxidation rates were consistent for wild-type hephaestin and each of the mutants (Tables 3, S2). Thus, these two parameters were closely monitored while analyzing the enzymatic activity of rhHp and the mutants of the putative iron ligands.
Ferroxidation rates of rhHp and mutants were studied in the range of 2–100 μM iron. Although the kinetic curves of IB, IB4, and IB2 are linear within this range of iron concentrations, the initial velocity versus substrate plots for wild-type hephaestin and the IB6 mutant show distinguishable hyperbolae at low iron concentrations (Fig. 5, inserts). This observation is also supported by the Eadie–Hofstee plots, which resemble characteristic biphasic curves only for rhHp and IB6; the remaining mutants produce linear Eadie–Hofstee plots with only the low-affinity component present (Fig. 5). Thus, the only mutant that retained a high-affinity iron oxidation component was IB6—the mutant with an unaffected iron-binding site in domain 6.

Ferroxidase activity of rhHp and mutants. Representative Eadie–Hofstee plots are shown, with the corresponding kinetic curves in the inserts: a rhHp, b IB6, c IB4, d IB2, and e IB
Discussion
Using separation by IEC, we purified rhHp with improved catalytic and spectroscopic properties. When determined under similar conditions, the k cat for Fe2+ of rhHp (18 min−1) was higher than ferroxidation rates previously reported for hephaestin (0.74 min−1 [14], 2.5 min−1 [17]). However, IEC-fractionated rhHp gave an absorbance coefficient at 600 nm (ε 600 nm = 5,500 M−1 cm−1) that indicated the presence of only one type 1 copper per molecule of hephaestin. These data disagree with the structural model of hephaestin ectodomain based upon ceruloplasmin [10] that predicted three type 1 copper sites. Furthermore, the total copper content of hephaestin isolated in this study is 3.5 atoms per molecule rather than the predicted six atoms. There are several possible explanations for the lower copper content of rhHp. Because the tissue culture medium was supplemented with 10 μM CuSO4, it is unlikely that the availability of copper was limiting during BHK cell growth. However, it is possible that there was incomplete copper loading of hephaestin during its biosynthesis by a heterologous expression system. It is also possible that hephaestin may contain only four copper atoms arranged as a single type 1 copper and a trinuclear cluster—this in itself would be sufficient to give ferroxidase activity as in the yeast ferroxidase Fet3p [26]. In this case, hephaestin is most likely to retain the type 1 copper atom in domain 6, which is adjacent to the trinuclear cluster. This suggestion is supported by the presence of a functional iron-binding site in domain 6 of human hephaestin (see later). Domains 2 and 4 of hephaestin, which also contain predicted type 1 copper ligands, may still play important roles in maintaining the overall structure of the molecule or may participate in the interactions of hephaestin with other proteins.
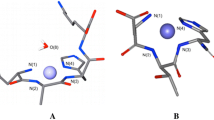
Detailed kinetic analysis of rhHp ferroxidation rates revealed non-Michaelis–Menten kinetics. When the initial kinetic data were plotted in Eadie–Hofstee coordinates, the plot displayed a biphasic profile, suggesting the presence of both low-affinity and high-affinity iron-binding sites (Fig. 4b). Similar biphasic Eadie–Hofstee plots were generated when the ferroxidase activity of ceruloplasmin was analyzed [27–29]. Furthermore, equilibrium dialysis studies have shown that ceruloplasmin possesses iron-binding sites with different affinities [30]. In ceruloplasmin, the predicted high-affinity Fe(II) binding sites are located beneath large protein loops [12, 30, 31] (Fig. 6). Owing to the abundance of acidic amino acid residues, the high-affinity iron-binding sites and the surrounding protein surface harbor significant negative charge [12] (Fig. 6c). This negatively charged area on the upper side of the ceruloplasmin molecule may also be responsible for low-affinity iron binding. The overall structure of the molecule as well as the charge distribution in ceruloplasmin was predicted to be conserved in hephaestin [10]. Together with the results of kinetic analysis, these data suggest that hephaestin and ceruloplasmin use a similar mechanism for iron oxidation.

Iron binding by human ceruloplasmin. a Surface model of ceruloplasmin; predicted ligands of high-affinity iron-binding sites in domain 4 (E597, H602, D684, E971) and in domain 6 (E935, H940, D1025, E272) are shown as sticks. b Same as a; protein loops which cover iron-binding sites are represented by ribbons; iron-binding sites in domain 4 (at the bottom) and domain 6 (at the top) are encircled. c Surface charge distribution on human ceruloplasmin. The negative and positive potential regions are scaled from red for −76 to blue for +76. The crystal structure of ceruloplasmin (Protein Data Bank code 2J5W) was visualized with PyMOL; the electrostatic map was obtained using the APBS plug-in
To study the role of the predicted iron ligands in ferroxidase activity of rhHp, we produced a mutant with all three putative iron-binding sites affected (IB) and mutants that retained a single predicted site for Fe(II) binding (IB2, IB4, IB6). Mutation of the amino acid residues in all three iron-binding sites resulted in a loss of high-affinity Fe(II) oxidation along with a sevenfold increase in the ratio between pPD- and Fe(II)-oxidation rates (Table 3, column 6). Similar results were obtained for the IB2 and IB4 mutants. In contrast, the IB6 mutant showed high-affinity iron oxidation with a K m comparable to that of wild-type rhHp (Table 3, column 4); the ratios of the pPD-and Fe(II)-oxidation rates for IB6 and rhHp were also equivalent (Table 3, column 6). These results are consistent with residues E960/H965 serving as iron ligands of the high-affinity binding site in domain 6 of rhHp. In contrast, residues E264/H269 in domain 2 and D616/H621 in domain 4 appear to be dispensable for hephaestin ferroxidase activity; supporting this observation, mutations of these residues in the IB6 mutant did not result in any detectable impairment of ferroxidase function. There is a possibility that the role of residues E264/H269 and D616/H621 in iron oxidation was not revealed owing to the absence of copper at type 1 sites in domains 2 and 4 of rhHp. On the other hand, iron-binding sites in domains 2 and 4 of rhHp are not composed of a canonical set of ligands. Conserved ferrous-binding sites in domain 6 of rhHp and in domains 4 and 6 of ceruloplasmin are formed by three acidic residues and a histidine residue. In contrast, the putative sites for iron binding in domains 2 and 4 of hephaestin have one of the acidic residues substituted with a serine residue. This unusual arrangement of ligands may have a detrimental effect on iron oxidation at these sites.
In conclusion, this work advances our current understanding of the catalytic mechanism of human hephaestin, one of three human multicopper ferroxidases [32]. The amino acid sequence homology between the ferroxidases predicts similar biochemical properties; however, any specific functions for each of the paralogs probably result from their structural differences. The current work on hephaestin has revealed substrate specificity, copper loading, and the ferroxidation mechanism in this multicopper ferroxidase. The knowledge acquired in the current study will facilitate further research focused on the structure and catalytic mechanism of iron oxidation by the ferroxidase paralogs.
Abbreviations
- BHK:
-
Baby hamster kidney
- DEAE:
-
Diethylaminoethyl
- DMEM-F12:
-
Dulbecco’s modified Eagle’s medium–Ham F12 nutrient mixture
- IEC:
-
Ion exchange chromatography
- pPD:
-
p-Phenylenediamine
- rhHp:
-
Recombinant human hephaestin
- Tris:
-
Tris(hydroxymethyl)aminomethane
References
Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ (1999) Nat Genet 21:195–199
Frazer DM, Vulpe CD, McKie AT, Wilkins SJ, Trinder D, Cleghorn GJ, Anderson GJ (2001) Am J Physiol Gastrointest Liver Physiol 281:G931–G939
Bleackley MR, Wong AY, Hudson DM, Wu CH, Macgillivray RT (2009) Transfus Med Rev 23:103–123
Aisen P, Leibman A, Zweier J (1978) J Biol Chem 253:1930–1937
Chen H, Attieh ZK, Su T, Syed BA, Gao H, Alaeddine RM, Fox TC, Usta J, Naylor CE, Evans RW, McKie AT, Anderson GJ, Vulpe CD (2004) Blood 103:3933–3939
Edwards JA, Bannerman RM (1970) J Clin Invest 49:1869–1871
Qian ZM, Chang YZ, Leung G, Du JR, Zhu L, Wang Q, Niu L, Xu YJ, Yang L, Ho KP, Ke Y (2007) Biochim Biophys Acta 1772:527–532
Hudson DM, Curtis SB, Smith VC, Griffiths TA, Wong AY, Scudamore CH, Buchan AM, MacGillivray RT (2010) Am J Physiol Gastrointest Liver Physiol 298:G425–G432
Pierre JL, Fontecave M (1999) Biometals 12:195–199
Syed BA, Beaumont NJ, Patel A, Naylor CE, Bayele HK, Joannou CL, Rowe PS, Evans RW, Srai SK (2002) Protein Eng 15:205–214
Sakurai T, Kataoka K (2007) Chem Rec 7:220–229
Lindley PF, Card G, Zaitseva I, Zaitsev V, Reinhammar B, Selin-Lindgren E, Yoshida K (1997) J Biol Inorg Chem 2:454–463
Machonkin TE, Zhang HH, Hedman B, Hodgson KO, Solomon EI (1998) Biochemistry 37:9570–9578
Vashchenko G, Bleackley MR, Griffiths TA, MacGillivray RT (2011) Arch Biochem Biophys 514:50–56
Young SN, Curzon G (1972) Biochem J 129:273–283
Zaitsev VN, Zaitseva I, Papiz M, Lindley PF (1999) J Biol Inorg Chem 4:579–587
Griffiths TA, Mauk AG, MacGillivray RT (2005) Biochemistry 44:14725–14731
Palmiter RD, Behringer RR, Quaife CJ, Maxwell F, Maxwell IH, Brinster RL (1987) Cell 50:435–443
Kammann M, Laufs J, Schell J, Gronenborn B (1989) Nucleic Acids Res 17:5404
Molday RS, MacKenzie D (1983) Biochemistry 22:653–660
Cuatrecasas P (1970) J Biol Chem 245:3059–3065
Stookey LL (1970) Anal Chem 42:779–781
Curzon G (1961) Biochem J 79:656–663
Rice EW (1962) Anal Biochem 3:452–456
Musci G, Di Marco S, Bellenchi GC, Calabrese L (1996) J Biol Chem 271:1972–1978
Taylor AB, Stoj CS, Ziegler L, Kosman DJ, Hart PJ (2005) Proc Natl Acad Sci USA 102:15459–15464
Osaki S (1966) J Biol Chem 241:5053–5059
Huber CT, Frieden E (1970) J Biol Chem 245:3973–3978
Ryan TP, Grover TA, Aust SD (1992) Arch Biochem Biophys 293:1–8
Brown MA, Stenberg LM, Mauk AG (2002) FEBS Lett 520:8–12
Quintanar L, Gebhard M, Wang TP, Kosman DJ, Solomon EI (2004) J Am Chem Soc 126:6579–6589
Chen H, Attieh ZK, Syed BA, Kuo YM, Stevens V, Fuqua BK, Andersen HS, Naylor CE, Evans RW, Gambling L, Danzeisen R, Bacouri-Haidar M, Usta J, Vulpe CD, McArdle HJ (2010) J Nutr 140:1728–1735
Acknowledgments
We thank Grant Mauk for many fruitful discussions. This work was supported in part by a grant from the Canadian Institutes of Health Research (to R.T.A.M.).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Vashchenko, G., MacGillivray, R.T.A. Functional role of the putative iron ligands in the ferroxidase activity of recombinant human hephaestin. J Biol Inorg Chem 17, 1187–1195 (2012). https://doi.org/10.1007/s00775-012-0932-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-012-0932-x