
Abstract
Metallothioneins (MTs) are a class of ubiquitously occurring low molecular mass, cysteine- and metal-rich proteins containing sulfur-based metal clusters formed with Zn(II), Cd(II), and Cu(I) ions. In mammals, four distinct MT isoforms designated MT-1 through MT-4 exist. The first discovered MT-1/MT-2 are widely expressed isoforms, whose biosynthesis is inducible by a wide range of stimuli, including metals, drugs, and inflammatory mediators. In contrast, MT-3 and MT-4 are noninducible proteins, with their expression primarily confined to the central nervous system and certain squamous epithelia, respectively. MT-1 through MT-3 have been reported to be secreted, suggesting that they may play different biological roles in the intracellular and extracellular space. Recent reports established that these isoforms play an important protective role in brain injury and metal-linked neurodegenerative diseases. In the postgenomic era, it is becoming increasingly clear that MTs fulfill multiple functions, including the involvement in zinc and copper homeostasis, protection against heavy metal toxicity, and oxidative damage. All mammalian MTs are monomeric proteins, containing two metal–thiolate clusters. In this review, after a brief summary of the historical milestones of the MT-1/MT-2 research, the recent advances in the structure, chemistry, and biological function of MT-3 and MT-4 are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In mammals, four distinct metallothionein (MT) isoforms designated MT-1 through MT-4 exist. The first MT, later found to be composed of the MT-1 and MT-2 isoforms, was discovered more than 50 years ago by Margoshes and Vallee [1] followed by the discovery of MT-3 [2] and MT-4 [3] at the beginning of 1990s. The most predominant isoforms MT-1/MT-2 are expressed in almost all tissues and their biosynthesis is inducible by a variety of stress conditions and compounds, including glucocorticoids, cytokines, reactive oxygen species (ROS), and metal ions [4]. In contrast, MT-3 and MT-4 are relatively unresponsive to these inducers and their expression is primarily confined to the central nervous system (CNS) [5] and cornified and stratified squamous epithelium [3], respectively. Presently, there are a great number of reviews covering different biological and structural aspects of MT research and focusing predominantly on MT-1/MT-2 [6–16]. In recent years the role of MTs in the brain, in particular of the MT-3 isoform, has been the subject of intense research. The biological studies on brain MTs are reviewed in this special issue. In this contribution, we briefly summarize the historical milestones of MT-1/MT-2 research till about 1990 and focus on the recent advances in the structure, chemistry, and biological functions of MT-3 and MT-4.
Metallothionein-1/metallothionein-2: historical highlights
As discussed by the late Bert L. Vallee [17] in his contribution to the first MT meeting, the discovery of MT has been closely linked with the search for biological material containing cadmium. The first report on the presence of this metal in human organs, in particular kidney, was in the Russian literature in 1941 [18]. In the mid-1950s the translated article became available to Western scientists. Upon screening for cadmium content in kidneys of a few mammalian species, Margoshes and Vallee [1] identified protein containing besides cadmium also a substantial amount of zinc and lesser amounts of copper and iron after fractionation of horse kidney cortex. The purification and characterization of this material by Kägi and Vallee [19] revealed a low molecular mass 6–7-kDa metal- and cysteine-rich protein, named “metallothionein” (MT) to reflect the extremely high thiolate sulfur and metal content, both of the order of 10% (w/w). Further chromatographic separation revealed the presence of two principal MTs, later designated as MT-1 and MT-2, of 61–62 amino acids, of which 20 were cysteines and none of which were aromatic amino acids. Both MTs contained seven Zn(II) or Cd(II)/Zn(II) ions. The subsequent protein sequencing showed a single polypeptide chain with distinct clustering of the 20 cysteine residues into Cys–X–Cys, Cys–Cys and Cys–X–X–Cys sequences, where X stands for amino acids other than cysteine [20] (Table 1). These unique features of the classical mammalian MT-1 and MT-2 isoforms attracted the interest of scientists from fields as different as biochemistry, molecular biology, toxicology, and structural biology.
The toxicology studies were initiated by the finding that MT was present in increased amounts in livers of rabbits exposed to cadmium. It has been postulated that this observation reflects the induced biosynthesis of this protein by this metal and its role in metal detoxification [22]. This initial assumption of MT-1/MT-2 induction by heavy metals was later confirmed by the increased MT content in liver, kidney, and intestines following the parenteral or dietary administration of cadmium, copper, or zinc to experimental animals [23]. In the years since this publication, a large number of articles on the role of MT-1/MT-2 in metal toxicity have been published and similar conclusions have been drawn. However, the real advances were the observations that mice that cannot synthesize any MT are sensitive to cadmium toxicity, whereas mice that express excess amounts of any MT are resistant to this metal [24–26]. It has been demonstrated, moreover, that selection for cadmium resistance with mammalian cells always results in up to 80-fold increase of the entire MT locus [27]. These observations reflected the original discovery of cadmium-containing MT in horse kidney [1]. Even though cases of cadmium toxicity are known, they are rare and exclusively caused by man. Therefore, it has been suggested that even though a characteristic phenotype of cells and mice with altered expression of MTs is the sensitivity to cadmium toxicity, it seems unlikely that the evolutionary conservation of these ubiquitous, inducible genes in most organisms is driven by the ability of MTs to detoxify cadmium [28]. Thus, the current notion is that cadmium detoxification is a property of MTs rather than its evolutionary function [29].
Molecular biology studies on MTs aimed at the understanding of the gene structure, function, and regulation. The genes for human and mouse MT-1/MT-2 have been most extensively studied [30–33]. Whereas four MT genes are present in the mouse genome, in human a significant genetic polymorphism exists. The human MT genes are located on band 13 of the long arm of chromosome 16 and are encoded by a multigene cluster of tightly linked genes. There are at least seven functional MT-1 genes and a single gene encoding the other MT isoforms MT-2 (MT-2A), MT-3, and MT-4 [13]. A number of other MT or MT-like genes and pseudogenes with significant homology to functional MT genes exist within the human genome, but their functionality is unknown. The MT genes encode for two-domain proteins. The three exons composing the human MT genes sequentially encode for the N-terminal region of the β-domain (exon 1), the rest of the β-domain (from residue 11 or 12; exon 2), and all of the α-domain (from residue 31 or 32; exon 3). Exons 2 and 3 are spliced at the junction of codons for the Lys/Lys or Arg/Lys residues in the interdomain region. The promoter regions of MT-1 and MT-2 genes contain several metal-responsive elements (MREs) and glucocorticoid-responsive elements as well as elements involved in basal level transcription. The MT-1/MT-2 expression is also regulated by oxidative stress by antioxidant responsive elements or by MREs that are also responsive to oxidants. Metal-regulatory transcription factor 1 (MTF-1), which is essential for basal expression of MT-1/MT-2 and induction by zinc, binds to promoter proximal MREs. MTF-1 binds to MREs through its six C2H2 zinc fingers [34]. Although similar MREs were also identified in the MT-3 gene, there were unresponsive to zinc [35]. The DNA sequences responsible for cell-specific regulation of MT-3 and MT-4 genes are currently unknown. The regulation of tissue-specific MT-3 gene expression in humans does not appear to involve a repressor. Thus, other mechanisms such as chromatin organization and epigenetic modifications may account for the presence or absence of MT-3 transcription.
In early biochemical studies the biological role of MT-1/MT-2 was sought. As the major zinc-binding proteins in the cell, it has long been hypothesized that MTs can potentially modulate many important biological processes that involve zinc-requiring enzymes. Their involvement can either be direct, via interaction with inactive apoenzymes, or indirect, by regulation of available Zn(II) in the cell. The first demonstration that several zinc-requiring apoenzymes can be reactivated by the transfer of Zn(II) from Zn7MTs in vitro was 30 years ago [36]. Evidence that apoMT, or thionein, readily removes Zn(II) from zinc-finger transcription factors such as Sp1 and Xenopus laevis TFIIIA in vitro, eliminating their DNA binding competence, has also been provided [37, 38]. The in vitro demonstration that MTs are extraordinarily efficient scavengers of free hydroxyl radicals formed during various forms of oxidative stress suggested that these proteins may be a part of such a protective system [39]. The primary targets of the free-radical attack in MTs are thiolate groups of the metal–thiolate clusters, which may be repaired in the cell by reduced glutathione. Following this discovery, evidence supporting such a physiological role of MTs in the cellular defense system against oxidative stress and their induced biosynthesis under these conditions was provided [40]. In the postgenomic era it became clear that MTs can have multiple biological functions.
In view of the high cysteine and metal content, the structural investigations of MT-1/MT-2 were highly challenging. Since most of these studies were conducted before 1990, no recombinant protein expression systems were available. The native MTs, as isolated from livers of experimental animals, contained either Zn(II) or both Zn(II) and Cd(II) ions. To examine the geometry and the organization of metal binding sites, the native metal ions were replaced by paramagnetic Co(II) or by the NMR-active 113Cd isotope (I = 1/2). The optical and magnetooptical (magnetic circular dichroism) studies of Co7MT provided evidence for a tetrahedral-type of geometry of all metal binding sites and their coordination by cysteine thiolates [41]. Convincing evidence for the two-cluster arrangement of the seven divalent metal ions in MT, i.e., for a three-metal and a four-metal cluster, has been provided by homonuclear 113Cd NMR decoupling studies of 113Cd7MT. In these studies, the Cd(II) coordination by both terminal and bridging thiolate ligands was inferred from the chemical shift position of the 113Cd resonances [42]. The observation of an antiferromagnetic coupling between metal centers in electron spin resonance and magnetic susceptibility measurements of Co7MT lend further support for a cluster structure in MTs [43]. Through enzymatic cleavage of Cd7MT and the 113Cd NMR characterization of the Cd4-containing peptide, the indication for a two-domain structure, each domain encompassing a metal-thiolate cluster, was obtained [44]. The mechanism of metal binding to apoMT-2 was studied by 1H NMR and magnetic susceptibility measurements of the paramagnetic Co(II) metalloform of the protein at room temperature. The results revealed a detailed binding scheme in which both metal–thiolate clusters in Co7MT-2 were formed in a noncooperative manner [45]. However, in similar studies using Cd(II), a specific cooperative binding to the α-domain followed by the β-domain was revealed by circular dichroism, magnetic circular dichroism, and 113Cd NMR measurements [46, 47]. The substantially different metal sizes of Cd(II) and Co(II), the latter being closely similar to that of Zn(II), thiolate affinities, and the corresponding bond lengths may account for the differences in the pathway of cluster formation.
Since early attempts to crystallize MT-1/MT-2 failed in a number of crystallography groups, a new solution NMR method for the 3D structure determination, developed by Kurt Wüthrich and coworkers in Zurich, was applied. The first structural studies of Wüthrich’s group were conducted on Cd7MT-2 from rabbit liver. However, the structure elucidation by NMR measurements turned out to be more difficult than expected because of the heterogeneity of native protein, the absence of aromatics, leading to very poor chemical shift dispersion, and the presence of Cd–Cys coordinative bonds in the clusters which could not be analyzed with nuclear Overhasuer effects. Crucial to the structure elucidation was the preparation of structurally homogeneous 113Cd7MT-2 samples, generated through the method of metal reconstitution, and the development of heteronuclear 1H–113Cd correlation experiments enabling the cadmium–cysteine topology of the clusters in the rabbit protein to be unraveled [48].
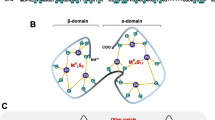
At about the time of the publication of the cluster topology in rabbit 113Cd7MT-2, the crystal structure of native rat Cd5,Zn2MT-2 was reported [49]. However, the sequence-specific metal-cysteine coordinative bonds in the crystal structure of the rat protein were substantially different from those found by NMR measurements for the rabbit protein. Given the amount of this covalent connectivity information, it was quite clear that this discrepancy between the two sets of results was not something that could be explained easily. Among others, the most likely reasons considered by the scientific community were (1) the NMR and crystallography results might both be correct as differences in the respective primary structures exist, i.e., the rat protein shows ten substitutions of non-cysteine amino acids, (2) the structural differences were caused by the metal reconstitution of the rabbit protein, and (3) the results obtained using the newly developed NMR technique are presumably incorrect. It is hard to imagine a less desirable event at the early stage in the development of the methodology for NMR structure determination of proteins than that the NMR method should produce a high-profile mistake. Also the structure of 113Cd7MT was one of the first protein structures determined by NMR measurements, and it was the first structure of a metalloprotein to be determined. In an effort to resolve the discrepancy, NMR studies aiming at the cluster topology in metal-reconstituted rat 113Cd7MT-2 and native rat 113Cd5,Zn2MT-2, generated by the administration of 113Cd(II) salt to experimental animals similarly to samples used in crystallization, were undertaken [50]. The NMR data obtained for the rat protein confirmed the cluster topology seen already with the rabbit protein (Fig. 1). Subsequently the NMR structures of 113Cd7MT-2 from rat and rabbit livers were completed [51, 52] and those of human 113Cd7MT-2, and, for comparison, native Zn7MT-2 were also determined [53, 54]. The NMR structures of these three homologous MTs are virtually identical, and later the crystal structure of rat Cd5,Zn2MT-2 was redetermined and found to coincide very closely with the NMR structure (Fig. 2) [55, 56].

Structure of the two metal–thiolate clusters with divalent metal ions (gray spheres) in mammalian MII 7MT-2. The sulfur atoms (yellow spheres) of cysteine thiolates are labeled with the cysteine residue number

The three-dimensional X-ray crystal structure of rat Zn2Cd5MT-2 (top) [55] and the NMR solution structure of the α-domain and β-domain of rat 113Cd7MT-2 (bottom) [51]. The Cd(II) and Zn(II) ions are shown as light-orange spheres and blue spheres, respectively, connected to the protein backbone by cysteine thiolate ligands. The models were generated with PyMOL using the Protein Data Bank coordinates of 4MT2, 1MRT, and 2MRT
The determination of the structure of mammalian and later also of the crustacean and echinoderm MTs established the presence of trinuclear and tetranuclear thiolate clusters with divalent metal ions as the characteristic structural motif. These structural studies led to the notion about this class of proteins which include the exclusive metal coordination by cysteines, the absence of aromatic amino acids (with rare exceptions) and secondary structure. However, in recent years the biochemical characterization of bacterial and plant MTs, which differ considerably from mammalian proteins, revealed that besides high cysteine content, also aromatic amino acids, including histidine, are present. Furthermore, structural studies on these MTs revealed that apart from the trinuclear and tetranuclear clusters, also mononuclear and binuclear metal binding sites with divalent metal ions occur. In addition, in these structures a well-defined protein fold and, in some instances, metal coordination by both cysteine and histidine residues are encountered. The structural and functional properties of bacterial and plant MTs are reviewed in this special issue.
Metallothionein-3
Biological properties of metallothionein-3
In the human brain the MT-1/MT-2 and MT-3 isoforms occur [9]. The most distinctive biological property of MT-3 is its extracellular growth inhibitory activity in neuronal primary cultures, which led to its discovery [5]. Thus, MT-3, but not MT-1/MT-2 antagonizes the ability of Alzheimer disease brain extract to stimulate survival and neuritic sprouting of cultured neurons [5, 57]. The observation that constitutive expression of MT-3 but not MT-1 inhibited the growth of cultured kidney cells under zinc-deficient conditions supports distinct functions of both isoforms [58]. The discovered bioactivity led to the hypothesis that MT-3 may be involved in pathogenic processes leading to Alzheimer disease. Independent studies supporting its role in Alzheimer disease showed that MT-3 but not MT-1/MT-2 protects neuronal cells from the toxic effect of amyloid-β (Aβ) peptide Aβ1–40 [59]. However, these two effects are functionally unrelated (see below).
That MT-3 displays biological properties not observed for MT-1/MT-2 is clearly documented by in vivo studies in which mice overexpressing MT-3 in most organs died as a result of pancreatic atrophy, whereas expression of similar amounts of MT-1 had no effect [60]. Although the reason for MT-3 toxicity is unknown, these results provide biological evidence that the MT isoforms have different functional properties. Other in vivo studies supporting this conclusion showed that in a mouse model of brain injury, exogenously administered MT-3, in contrast to human MT-2, does not affect inflammation, oxidative stress, and apoptosis [61]. The latter results highlight specific and divergent roles of exogenous MT-3 when compared with the MT-1/MT-2 isoforms.
Numerous studies investigated the localization of MT-1/MT-2 and MT-3 in the CNS. MT-1 and MT-2 are present throughout the brain and spinal cord, with the astrocytes being the main cell type expressing both isoforms. In contrast, MT-3, the isoform predominantly expressed in the CNS [5], is localized in both neurons and astrocytes [10, 62, 63], where it appears to play an important role in the homeostasis of copper and zinc [9]. The MT-1/MT-2 and MT-3 isoforms have been reported to be secreted, suggesting that they may play different biological roles in the intracellular and extracellular space [64–66]. Previously, the involvement of the receptor megalin in the renal uptake of MT-1/MT-2 was described [67]. The same receptor also mediates the transport of MT-1/MT-2 into neurons following brain injury [66]. Presently, no information on whether the megalin receptor also mediates the transport of MT-3 is available.
MT-3 is present in several brain regions at different concentrations, being particularly concentrated in presynaptic terminals of zinc-enriched neurons of cerebral cortex and hippocampus. Zinc-enriched neurons belong to a subset of glutamatergic neurons and as such contain presynaptic zinc vesicles in which 10–15% of the total Zn(II) in the brain is present. The colocalization of MT-3 and zinc vesicles in zinc-enriched neurons led to the suggestion that the protein may contribute to the utilization of Zn(II) as a neuromodulator [68]. Studies of Zn7MT-3 and zinc transporter 3 knockout mice revealed that zinc transporter 3, a transporter that concentrates Zn(II) in presynaptic vesicles, and MT-3 function in the same pathway [69]. From these studies an important role for MT-3 in the recycling of Zn(II) was suggested [70]. The demonstration of the direct interaction of Zn7MT-3 with Rab3A, a small GTPase involved in the regulation of the synaptic vesicle cycle, supports this role [71]. The immunochemical identification of MT-3 as a component of a brain multiprotein complex with heat shock protein 84 and creatine kinase [72] suggests that MT-3 may have additional intracellular functions that remain to be elucidated. Recently, the specific and reversible binding of one extra Zn(II) to Zn7MT-3 (K app ~100 μM), forming Zn8MT-3, was reported [73]. Since during synaptic signaling the free Zn(II) concentration can reach up to 300 μM [74], a reversible switch between the Zn7MT-3 and Zn8MT-3 forms may play a role as a zinc buffer or in an interaction with a binding partner(s). The analytical results of the latter study suggest that the reported coexistence of MT-3 species in solution with a metal distribution between five to nine M(II) per protein (M is Zn, Cd), detected by electrospray ionization (ESI) mass spectrometry (MS) upon binding of seven M(II) equivalents to apoMT-3, seems to reflect the reconstitution and ionization conditions used [75].
MT-3, like other MTs [39, 40, 76, 77], efficiently scavenges ROS. Thus, exogenous MT-3 prevents neurite extension and the death of differentiated cortical neurons caused by exposure to high oxygen concentrations, owing to its more efficient scavenging of free hydroxyl radicals than the same concentration of Zn7MT-1/MT-2 [64]. However, studies of MT-3-null mice following damage caused by a focal cryolesion onto the cortex showed that, in contrast to MT-1/MT-2, MT-3 is unlikely to be a significant factor for controlling the inflammatory response, oxidative stress, and apoptosis after significant brain damage, but it may influence neuronal regeneration [78]. The in vitro reactions of S-nitrosothiols and H2O2 with Zn7MT-1/MT-2 and Zn7MT-3 revealed that whereas Zn7MT-3 was significantly more reactive than Zn7MT-1/MT-2 toward S-nitrosothiols, the reactivity of all three isoforms toward H2O2 was comparable. The increased reactivity of Zn7MT-3 with free NO and S-nitrosothiols led to the proposal that Zn7MT-3 may specifically convert NO signals to zinc signals [79]. The metal-induced aggregation of Aβ and accompanied oxidative stress contribute to the progression of Alzheimer disease (see below). In vitro studies showed that in contrast to amorphous aggregates of Aβ formed upon Zn(II) addition, the Zn(II) release from Zn7MT-3 upon a slow cysteine oxidation by H2O2 induces fibrillar-type Aβ aggregates, thereby modulating its morphology. Thus, the protein may protect cells not only by its capacity for ROS scavenging, but also through Zn(II) release and subsequent binding to Aβ [80]. Recently, in view of the finding that MT-3 regulates lysosomal functions, possible roles of labile zinc and Zn7MT-3 in oxidative-stress-induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes have been described [81]. Given the accumulating evidence that lysosomes play key roles in many neurological diseases, the role of labile zinc and Zn7MT-3 in lysosomal function warrants further study.
As discussed above, the regulation of the basal MT-3 expression is poorly understood. Nevertheless, in recent years overexpression of MT-3 in certain cancer types including prostate [82] and breast [83] has been described. Since the MT-3 gene was identified as hypoxia-inducible in several human tissues [84], the overexpression of MT-3 observed in several cancers presumably reflects the hypoxic conditions, due to insufficient vascularization of the fast-proliferating cancer tissue. MT-3, similarly to MT-1/MT-2, may confer cellular resistance in platinum-based chemotherapy in these cell types [85]. However, kinetics of the reaction of cisplatin with Zn7MT-3 and Zn7MT-2 revealed a substantially higher reactivity of cisplatin toward Zn7MT-3 than Zn7MT-2 [86].
Structure of metallothionein-3 with divalent metal ions
The distinct biological activity of MT-3 has been linked to the structural differences between MT-1/MT-2 and MT-3. As the structure of MTs is determined by the interplay between the polypeptide chain and metal ions, the differences in amino acid sequences ought to be responsible for the bioactivity. Compared with the amino acid sequences of mammalian MT-1/MT-2 (Table 1), the MT-3 sequence shows two inserts, an acidic hexapeptide in the C-terminal region and a threonine in position 5 in the N-terminal region followed by a conserved C6PCP9 motif. Structural information on the metal–thiolate clusters in MII 7MT-3 was forthcoming from spectroscopic investigations of the recombinant protein and chemically synthesized single protein domains. The studies revealed the presence of two mutually interacting protein domains, with each domain encompassing a metal–thiolate cluster [87–90]. A three-metal cluster is located in the N-terminal β-domain (residues 1–31) and a four-metal cluster is located in the C-terminal α-domain (residues 32–68) of MII 7MT-3 [88, 89], a metal organization found also in MII 7MT-1/MT-2 (Fig. 2). From 113Cd NMR studies of human 113Cd7MT-3, evidence for unprecedented dynamic processes within the metal–thiolate clusters was obtained [90]. From significant broadening of all 113Cd resonances and the very low and temperature-independent intensity of the Cd3Cys9 cluster resonances, the presence of dynamic processes acting on two different 113Cd NMR timescales was suggested: (1) fast exchange between conformational cluster substates giving rise to broad, weight-averaged resonances and (2) additional very slow exchange processes between configurational cluster substates in the β-domain. The changes in conformational substates may be visualized as minor dynamic fluctuations of the metal coordination environment and those of the configurational substates as major structural alterations brought about by temporarily breaking and reforming of the metal-thiolate bonds [90]. The existence of interchanging configurational cluster substates of comparable stability was demonstrated for inorganic adamantane-like metal–thiolate clusters with the general formula [M4(SPh)10]2− (M is Cd(II), Zn(II), Co(II), and Fe(II)] [91]. Studies toward obtaining the 3D NMR structure of mouse and human Cd7MT-3 have also been undertaken [92, 93]. However, because of dynamic processes in the β-domain of Cd7MT-3, only the 3D structure of the C-terminal α-domain (residues 32–68), containing an adamantane-like Cd4Cys11 cluster, could be determined by NMR measurements. The structure of this domain reveals a peptide fold and cluster organization very similar to that found in mammalian Cd7MT-1/MT-2, with the exception of an extended flexible loop encompassing the acidic hexapeptide insert (Fig. 3).

The NMR solution structure of the α-domain of human 113Cd7MT-3 [93]. The Cd(II) ions are shown as light-orange spheres connected to the protein backbone by cysteine thiolate ligands. The model was generated with PyMOL using the Protein Data Bank coordinates of 2F5H
To account for slow dynamic events centered at the three-metal cluster of MT-3, a partial unfolding of the β-domain, whose kinetics could be determined by the cis/trans interconversion of Cys-Pro amide bonds in the C6PCP9 motif, has been suggested [94]. Support for this model came from extended X-ray absorption fine structure (EXAFS) studies of Zn7MT-3 and its single Zn3β-domain where, besides evidence for the tetrahedral tetrathiolate Zn(II) coordination, also indication of an unusual structure of the three-metal cluster was obtained. In both Zn3β-domains a Zn···Zn backscattering at about 3.28 Å was detected (Fig. 4) [87]. Since this distance is inconsistent with the cyclohexane-like Zn3Cys9 cluster structure present in the β-domain of mammalian MT-2, where a Zn···Zn distance of about 3.8 Å exists [56], it may reflect a predominant population of one cluster configuration under the conditions used (at 77 K).

Structural model of the Zn3(Cys)9 cluster in Zn7MT-3 (right) and in comparison with the cyclohexane-like Zn3(Cys)9 cluster present in Zn7MT-2 (left). Metal ions are shown as blue spheres connected to cysteine sulfur ligands (yellow). EXAFS: extended X-ray absorption fine structure. (Adapted with permission from [87])
Additional insights into the dynamics of the β-domain of Cd7MT-3 were provided by molecular dynamics simulations. The simulation of the partial unfolding supported the proposed role of cis/trans interconversion of Cys-Pro amide bonds in the folding/unfolding process of the β-domain of Cd7MT-3 [95]. In other molecular dynamics simulations, a correlation between the number of backbone amide hydrogen bonds to metal coordinated cysteines, NH–Sγ hydrogen bonds, in mammalian Cd7MTs has been proposed to represent a controlling factor regulating the metal-thiolate cluster dynamics [96].
The C6PCP9 motif was the first segment found to be important for the extracellular growth inhibitory activity of Zn7MT-3 [57, 94]. Mutational studies on biologically inactive Zn7MT-1 revealed that besides the introduced C6PCP9 motif, also the unique Thr5 is required for the bioactivity and structure dynamics [97]. Further insights into the structural features underlying the bioactivity of Zn7MT-3 came from a number of mutational studies which revealed that other amino acids in the β-domain along with domain–domain interactions, mediated partly through the acidic hexapeptide insert in the α-domain, are important (reviewed in [98]). Thus, although the mechanisms underlining these biological effects remain to be elucidated, the results obtained so far suggest that the structure of the β-domain of Zn7MT-3 is subjected to a fine-tuning. Taken together, the findings of the structural and mutational studies led to the conclusion that both the specific structural features and the structure dynamics are necessary prerequisites for the extracellular biological activity of Zn7MT-3 [94, 97, 98]. The bioactivity in neuronal assays has been established for Cu(I)4Zn3–4MT-3 isolated from human [2] and bovine [99] brains and for recombinant human Zn7MT-3 [98, 100]. However, comparative biological studies on well-defined metalloforms are currently lacking. Recently, two reviews focusing on various aspects of the structure and biology of MT-3 have been published [101, 102].
Structural features of Cu(I)-bound metallothionein-3
Whereas isolated mammalian MT-1 and MT-2 from livers of various species usually contain seven Zn(II), MT-3 isolated from human and bovine brains contains four Cu(I) and between three and four Zn(II). The isolated Cu(I)4Zn3–4MT-3 is a monomeric protein stable in air. EXAFS studies on Cu(I)4Zn3–4MT-3 revealed the presence of two homometallic clusters, a Cu(I)4–thiolate cluster and a Zn3–4–thiolate cluster [87]. The EXAFS data indicated that in contrast to tetrahedrally coordinated Zn(II) ions, Cu(I) ions are diagonally and/or trigonally coordinated by two or three cysteine ligands [87]. This observation, also seen for the Cu(I)–thiolate clusters in MT-1/MT-2, signifies that there are different coordination geometries for the binding of monovalent and divalent metal ions to MT-3. Therefore, to accommodate metal–thiolate clusters with different coordinating geometries, the structure should possess a high degree of flexibility.
Detailed information on the interaction of Cu(I) with MT-3 was forthcoming from spectroscopic studies of Cu(I) binding to the metal-free α-domain and β-domain and the full-length protein. The stepwise Cu(I) binding to both individual MT-3 domains showed that two well-defined Cu(I)–thiolate cluster forms are generated during this process. In the case of the β-domain (residues 1–32; Table 1), the successive formation of two cluster forms involving all nine cysteine ligands was observed, i.e., Cu4S9 and Cu6S9 clusters [88]. Similar studies on the α-domain (residues 32–68), containing 11 cysteine ligands, resulted in the formation of a Cu4S8–9 cluster followed by a Cu6S11 cluster [89]. In addition, the formation of the Cu4S9 cluster in both the single β-domain and full-length protein was cooperative [88, 103]. The cooperative formation of a stable folding intermediate containing a Cu4S9 cluster in the β-domain has also been shown by ESI–MS [104]. The major differences in the respective spectroscopic features between the Cu(I)4 and Cu(I)6 cluster forms were observed in the low-temperature Cu(I) luminescence spectra at 77 K [88, 89, 103]. Whereas the Cu(I)6 cluster exhibited only a single emission band at 600 nm, in the case of the Cu(I)4 clusters two emission bands at 420 and 610 nm were discerned. The presence of two emission bands in Cu(I)4 clusters has been correlated with short intranuclear Cu···Cu distances (less than 2.8 Å), allowing metal–metal interactions due to a d 10–d 10 orbital overlap. Accordingly, the high-energy emission band has been assigned to a 3CC (cluster-centered) origin and that at low energy to triplet charge transfer [88, 89, 105]. The metal–metal interactions may contribute to the stability of the Cu(I)4–thiolate cluster to oxidation in air. This is supported by the observation that the expansion of the metal core, generating a Cu(I)6–thiolate cluster, results in increased Cu···Cu distances and susceptibility to oxidation [105]. Similarities between the luminescence spectra of isolated Cu4,Zn3–4MT-3 and the Cu(I)4–thiolate cluster in the single β-domain suggested the presence of this cluster also in the isolated protein. Taken together the data indicated that the β-domain of MT-3 shows clear preference for binding of Cu(I) over Zn(II) ions.
The generation of a nativelike MT-3 structure through direct Cu(I) and Zn(II) incorporation into the recombinant apoprotein revealed monomeric Cu4,Zn4MT-3, in which, besides the Cu(I)4 cluster in the β-domain, a Zn4 cluster in the α-domain was present [103]. Whereas the Cu(I)4 cluster in Cu4,Zn4MT-3 possesses the already mentioned stability to air oxygen, the Zn4 cluster was found to be air-sensitive. Its oxidation resulted in disulfide formation and the release of one Zn(II), yielding Cu4,Zn3MT-3. This process could be prevented or reversed under reducing conditions [106].
Roles of metallothionein-3 in metal-linked neurodegenerative disorders
A predisposing risk factor associated with neurodegenerative diseases is age. In the normal brain a high concentration of essential transition metal ions such as zinc, copper, and iron ions is present. Dysregulated metal homeostasis, abnormal metal–protein interactions, and the associated oxidative stress, protein misfolding, and aggregation are critical common pathological hallmarks of the progression of several metal-linked neurodegenerative disorders [107, 108]. Insoluble protein deposits and diffusible oligomers are composed of individual amyloidogenic proteins or peptides such as the Aβ peptides, a major component of extracellular amyloid plaques in Alzheimer disease, prion protein in prion deposits typical of Creutzfeldt–Jakob disease, α-synuclein (α-Syn) in intracellular Lewy bodies in Parkinson disease, superoxide dismutase (SOD-1) aggregates in amyotrophic lateral sclerosis, and Huntington inclusions in cases of Huntington disease. In these neurodegenerative diseases, the expression of MT-3 has been found to be downregulated or altered [109–111] and changes in normal homeostasis of essential transition metals such as zinc and copper have been implicated as possible etiological factors [108, 112]. In contrast to redox-inert zinc, redox-active copper aberrantly bound to amyloidogenic proteins can react with molecular oxygen, resulting in the production of ROS through Fenton and Haber–Weiss reactions [113]. That MT-3 may play an important role in the progression of these diseases has been reviewed [111].
A protective role of extracellular Zn7MT-3 from Cu(II) toxicity has been suggested on the basis of investigations of its reactivity toward free Cu(II) ions [114]. The results showed that Zn7MT-3 through Cu(II) reduction to Cu(I) by thiolate ligands and binding to the protein, forming an air-stable Cu(I)4Zn4MT-3 species, efficiently scavenges and redox-silences the free Cu(II) ions. In this reaction, a Cu(I)4–thiolate cluster is formed cooperatively in the β-domain of the protein concomitant with two intramolecular disulfide bonds and the release of three Zn(II) ions [114]. Since four Zn(II) ions remained bound, the presence of an intact Zn4 cluster in the α-domain was suggested. The formation of Cu(I)4Zn4MT-3 completely quenched the ascorbate-driven and Cu(II)-catalyzed production of ROS [114]. Taken together the findings indicate that two types of air-stable Cu(I)4–thiolate clusters can be formed in the more reactive β-domain of MT-3, i.e., a fully reduced Cu(I)4S9 cluster and a Cu(I)4S5+x cluster. In the latter, by analogy with inorganic model complexes, the participation of a disulfide bridge sulfur (indicated by x) besides thiolates in the Cu(I) coordination cannot be excluded.
Studies aimed at understanding the protective effect of human Zn7MT-3 against Aβ1–40 toxicity showed that the protein can efficiently remove copper not only from soluble Aβ1–40–Cu(II) oligomers, but also from insoluble aggregates. In this process, Cu(II) is reduced by protein thiolates, forming the stable Cu(I)4Zn4MT-3 species described above and the non-redox-active Aβ1–40–Zn(II). This metal swap completely quenches the ROS production mediated by Cu(II) bound to Aβ1–40 and occurs not only in vitro, but also in human neuroblastoma cell culture, whereby the toxic effect of Aβ1–40–Cu(II) is abolished [115]. In recent studies, the protective effect of human Zn7MT-2A against Aβ1−40–Cu(II) toxicity was also investigated and compared with that of Zn7MT-3 [116]. In neuronal cell culture, a similar protective effect has been shown. The results of the reaction between Aβ1−40–Cu(II) and Zn7MT-2A have been discussed together with the chemical data on the fully reduced Cu(I)10MT-2A and Cu(I)12MT-3 species, generated in the reaction of apoMT-2A or apoMT-3 with the Cu(I)-DTT complex under strictly reducing conditions. It has been suggested, moreover, that the increased affinity of MT-2A for Cu(I) over MT-3 makes MT-2A a better protecting agent [116]. However, as the reduction of Cu(II) in Aβ1–40–Cu(II) to Cu(I) is accomplished by the thiolate ligands of both MTs, the formation of fully reduced Cu(I)MT-2A/MT-3 metalloforms in the oxidizing extracellular environment is highly unlikely. That under these conditions the reaction between Aβ1−40–Cu(II) and Zn7MT-2A also leads to Cu(I)4Zn4MT-2A, in which a Cu(I)4–thiolate cluster and two disulfide bonds are present, has been revealed (G. Meloni, C.L. Seeland, and M. Vašák, unpublished). Note also that the implicated direct metal swap between Cu(II), coordinated by nitrogen and oxygen ligands in Aβ1–40–Cu(II), and Zn(II), coordinated by thiolate ligands in Zn7MT-2A, without preceding Cu(II) reduction cannot occur. Consequently, the differences in Cu(I) affinity for MTs are not essential for the protective effect, but rather the long-term stability of partially oxidized Cu(I),ZnMTs formed upon Cu(II) reduction and removal from Aβ1−40–Cu(II) in the oxidizing extracellular space are essential. The ensuing redox cycling of copper in oxygen-sensitive Cu(I),ZnMTs can change their properties from antioxidant to prooxidant. In this context it may be noted that although the MT-1/MT-2 isoforms have been found to be significantly upregulated in regions of Aβ plaques in the Alzheimer disease brain [117–120], the presence of substantial concentrations of Cu(II) in these plaques (0.3 mM) has been shown [108, 121]. Clearly, more studies regarding the stability of brain MTs formed in the reaction between Zn7MTs and different concentrations of Cu(II) to molecular oxygen are needed.
Besides Alzheimer disease, a protective effect of MT-3 has also been considered in other metal-linked neurodegenerative diseases such as Parkinson disease and prion diseases [111]. Studies conducted on hemiparkinsonian rats suggest that the free-radical scavenging potency, including that of MT-3, is reduced in the Parkinson disease brain [111]. In Parkinson disease, the fibrillation and aggregation of α-Syn is a key process in the formation of intracellular inclusions, Lewy bodies, in neurons of substantia nigra pars compacta [122]. Aberrant tight binding of one Cu(II) to α-Syn and associated oxidative stress appears to contribute to the degeneration of dopaminergic neurons through the abnormal aggregation of this protein [123–125]. In prion diseases, transmitted by proteinaceous infective agents (prions) [126], the transition from natively folded prion protein (PrPC) to misfolded prion protein (PrPSc) is a crucial pathogenic event [107, 127]. The mainly disordered part of the PrPC structure can bind up to six Cu(II) ions [107]. Also, in this case oxidative stress, associated with the copper-catalyzed transformations of prion protein, plays an important role in the disease progression. Recent in vitro studies into the role of Zn7MT-3 in Parkinson disease and prion diseases have shown that Zn7MT-3, through Cu(II) removal from α-Syn and prion protein and the formation of air-stable Cu(I)4Zn4MT-3 species, efficiently prevents the deleterious redox activity of these proteins [128]. In view of widely different Cu(II) binding motifs in Aβ, α-Syn, and prion proteins, a general protective role of Zn7MT-3 against Cu(II) toxicity in the brain can be envisaged.
Metallothionein-4
The last member of the mammalian MT gene family MT-4 is located about 20 kb upstream from the 5′ of the MT-3 gene in both the mouse and the human genome [3]. MT-4 consists of 62 amino acids, with an insert of glutamate in position 5 relative to the classical MT-1 and MT-2 proteins (Table 1). Murine MT-4 messenger RNA appears to be present exclusively in cornified and stratified squamous epithelia. Many of these epithelia develop parakeratosis during zinc deficiency in rats. Differentiation of stratified epithelium involves movement of keratinocytes out of the basal, proliferative layer into the overlaying region where synthesis of unique cytoskeletal proteins begins. In situ hybridization revealed intense labeling of MT-4 messenger RNA in the differentiating spinous layer of cornified epithelia, whereas MT-1 was expressed predominantly in the basal, proliferative layer. Thus, a switch in MT isoform synthesis occurs during differentiation. The MT-4 derived from tongue epithelium contained both zinc and copper (ratio 2.6:1) [3]. From molecular and cell biology studies, involvement of MT-4 in regulating zinc metabolism during the differentiation of stratified epithelia has been suggested [3]. Molecular biology and expression profile studies of MT-4 in mammalian maternal decidua [129] and during epithelia development and physiology [3, 130] revealed that the MT-4 gene is subject to a strict developmental regulation.
The function of MT-4 in handling divalent Zn(II) or monovalent Cu(I) metal ions has been inferred from its structural properties. The organization, stability, and assembly of divalent metal ions in mammalian MT-4 have been investigated using the Cd(II) and Co(II) metalloforms of the protein and have been compared with the well-characterized mammalian Cd7MT-2 [131]. Both 113Cd NMR studies of reconstituted 113Cd7MT-4 and spectroscopic characterization of Co7MT-4 revealed that, similarly to the classical MT-1 and MT-2 proteins, the seven divalent metal ions are organized into two independent Cd4Cys11 and Cd3Cys9 clusters, with each metal ion tetrahedrally coordinated by terminal and μ-bridging cysteine ligands [131]. The cluster formation in Cd7MT-4 was cooperative and sequential, with the Cd4Cys11 cluster in the α-domain being formed first. In addition, the metal–thiolate clusters in MT-4 appear more stable to demetallation by EDTA than those of MT-1 [132]. The decreased ligand substitution reactivity of EDTA observed with Cd7MT-4 presumably reflects marked differences in the cluster geometry in Cd7MT-4 compared with Cd7MT-1/MT-2 [131].
Insights into the Cu(I) binding specificity of MT-4 have been obtained from the characterization of metalloforms upon heterologous expression of MT-4 and its individual domains in Escherichia coli culture using zinc-, cadmium-, or copper-supplemented media and their comparison with the well-characterized metalloforms of MT-1 [133]. Spectroscopic and ESI–MS analyses of the metal composition of the purified full-length protein and the individual α- and β-domains revealed that compared with MT-1, the MT-4 isoform shows an increased preference for monovalent copper ions over divalent zinc or cadmium ions, properties reflected in the formation of mixed MII (M is Zn, Cd) and Cu(I) species [133]. On the basis of these results and the in silico protein sequence analyses, it has been concluded that MT-4 differentiated to a MT with a “copper thionein” character rather than to a MT with a “zinc thionein” character. It has been suggested, moreover, that the dissimilarities in non-cysteine residues between MT-4 and MT-1 are responsible for the differences in metal binding behavior. Furthermore, the diminished Cd(II) binding ability of Cd4-αMT-4 versus Cd4-αMT-1 has been attributed to its high proline content, in agreement with the association of Cys-Pro motifs to structural perturbations in MT-3 [97]. Overall, the structural studies support the role of MT-4 in both zinc and copper metabolism in epithelia. Nevertheless, more work regarding the 3D structure of MT-4 and its physiological role are needed.
In more recent studies, the presence of about 14 and 3 mol of sulfide (S2−) ligands in recombinant full-length CdMT-4 and CdMT-1, respectively, has been reported [134, 135]. This finding led to the reinterpretation of the all assumed heterometallic Zn,Cd-βMT-4 species in the previous heterologous expression of MT-4 [133] as Cd-βMT-4 complexes that include sulfide ligands. However, although in these studies convincing analytical evidence for the presence of sulfide in these and other recombinant preparations of mammalian MTs was provided, in the analyses for MT isoforms present in the cytosol of mammalian cells and tissues by the combination of various analytical techniques, including capillary electrophoresis (CE) inductively coupled plasma MS and CE–ESI–MS, mass peaks of fully metal occupied MT isoforms without sulfide have been reported [136, 137]. Since in these studies MT isoforms containing sulfide may have escaped detection owing to their substantially increased molecular masses, more studies are needed to clarify the presence of sulfide in mammalian MTs.
References
Margoshes M, Vallee BL (1957) J Am Chem Soc 79:4813–4814
Uchida Y, Takio K, Titani K, Ihara Y, Tomonaga M (1991) Neuron 7:337–347
Quaife CJ, Findley SD, Erickson JC, Froelick GJ, Kelly EJ, Zambrowicz BP, Palmiter RD (1994) Biochemistry 33:7250–7259
Miles AT, Hawksworth GM, Beattie JH, Rodilla V (2000) Crit Rev Biochem Mol Biol 35:35–70
Masters BA, Quaife CJ, Erickson JC, Kelly EJ, Froelick GJ, Zambrowicz BP, Brinster RL, Palmiter RD (1994) J Neurosci 14:5844–5857
Coyle P, Philcox JC, Carey LC, Rofe AM (2002) Cell Mol Life Sci 59:627–647
Duncan KER, Stillman MJ (2006) J Inorg Biochem 100:2101–2107
Stillman MJ (1995) Coord Chem Rev 144:461–511
Hidalgo J, Aschner M, Zatta P, Vašák M (2001) Brain Res Bull 55:133–145
West AK, Hidalgo J, Eddins D, Levin ED, Aschner M (2008) Neurotoxicology 29:489–503
Krezel A, Hao Q, Maret W (2007) Arch Biochem Biophys 463:188–200
Henkel G, Krebs B (2004) Chem Rev 104:801–824
Cherian MG, Jayasurya A, Bay BH (2003) Mutat Res 533:201–209
Theocharis SE, Margeli AP, Klijanienko JT, Kouraklis GP (2004) Histopathology 45:103–118
Vašák M, Hasler DW (2000) Curr Opin Chem Biol 4:177–183
Vašák M, Romero-Isart N (2005) In: King RB (ed) Encyclopedia of inorganic chemistry, 2nd edn. Wiley, New York, pp 3208–3221
Vallee BL (1979) Experientia Suppl 34:19–39
Maliuga DP (1941) Dokl Akad Nauk USSR 31(2):145
Kägi JHR, Vallee B (1960) J Biol Chem 235:3460–3465
Kojima Y, Berger C, Vallee BL, Kägi JHR (1976) Proc Natl Acad Sci USA 73:3413–3417
Gouy M, Guindon S, Gascuel O (2010) Mol Biol Evol 27:221–224
Piscator M (1964) Nordisk Hygienisk Tidskrift. XLV:76–82
Nordberg M, Kojima Y (1979) In: Kägi JHR, Nordberg M (eds) Metallothionein. Birkhäuser, Basel, pp 41–116
Michalska AE, Choo KHA (1993) Proc Natl Acad Sci USA 90:8088–8092
Masters BA, Kelly EJ, Quaife CJ, Brinster RL, Palmiter RD (1994) Proc Natl Acad Sci USA 91:584–588
Liu Y, Liu J, Iszard MB, Andrews GK, Palmiter RD, Klaassen CD (1995) Toxicol Appl Pharmacol 135:222–228
Durnam DM, Palmiter RD (1987) Experientia Suppl 52:457–463
Palmiter RD (1998) Proc Natl Acad Sci USA 95:8428–8430
Vallee BL (1987) Experientia Suppl 52:5–16
Searle PF, Davison BL, Stuart GW, Wilkie TM, Norstedt G, Palmiter RD (1984) Mol Cell Biol 4:1221–1230
Hamer DH (1986) Annu Rev Biochem 55:913–951
West AK, Stallings R, Hildebrand CE, Chiu R, Karin M, Richards RI (1990) Genomics 8:513–518
Palmiter RD, Findley SD, Whitmore TE, Durnam DM (1992) Proc Natl Acad Sci USA 89:6333–6337
Heuchel R, Radtke F, Georgiev O, Stark G, Aguet M, Schaffner W (1994) EMBO J 13:2870–2875
Chapman GA, Kay J, Kille P (1999) Biochim Biophys Acta 1445:321–329
Udom AO, Brady FO (1980) Biochem J 187:329–335
Zeng J, Heuchel R, Schaffner W, Kägi JH (1991) FEBS Lett 279:310–312
Zeng J, Vallee BL, Kägi JH (1991) Proc Natl Acad Sci USA 88:9984–9988
Thornalley PJ, Vašák M (1985) Biochim Biophys Acta 827:36–44
Sato M, Bremner I (1993) Free Radic Biol Med 14:325–337
Vašák M (1980) J Am Chem Soc 102:3953–3955
Otvos JD, Armitage IM (1980) Proc Natl Acad Sci USA 77:7094–7098
Vašák M, Kägi JH (1981) Proc Natl Acad Sci USA 78:6709–6713
Boulanger Y, Armitage IM, Miklossy KA, Winge DR (1982) J Biol Chem 257:13717–13719
Bertini I, Luchinat C, Messori L, Vašák M (1989) J Am Chem Soc 111:7296–7300
Stillman MJ, Zelazowski AJ (1988) J Biol Chem 263:6128–6133
Good M, Hollenstein R, Sadler PJ, Vašák M (1988) Biochemistry 27:7163–7166
Frey MH, Wagner G, Vašák M, Sorensen OW, Neuhaus D, Wörgötter E, Kägi JHR, Ernst RR, Wüthrich K (1985) J Am Chem Soc 107:6847–6851
Furey WF, Robbins AH, Clancy LL, Winge DR, Wang BC, Stout CD (1986) Science 231:704–710
Vašák M, Wörgötter E, Wagner G, Kägi JH, Wüthrich K (1987) J Mol Biol 196:711–719
Schultze P, Wörgötter E, Braun W, Wagner G, Vašák M, Kägi JH, Wüthrich K (1988) J Mol Biol 203:251–268
Arseniev A, Schultze P, Wörgötter E, Braun W, Wagner G, Vašák M, Kägi JHR, Wüthrich K (1988) J Mol Biol 201:637–657
Messerle BA, Schäffer A, Vašák M, Kägi JH, Wüthrich K (1990) J Mol Biol 214:765–779
Messerle BA, Schäffer A, Vašák M, Kägi JH, Wüthrich K (1992) J Mol Biol 225:433–443
Robbins AH, McRee DE, Williamson M, Collett SA, Xuong NH, Furey WF, Wang BC, Stout CD (1991) J Mol Biol 221:1269–1293
Braun W, Vašák M, Robbins AH, Stout CD, Wagner G, Kägi JH, Wüthrich K (1992) Proc Natl Acad Sci USA 89:10124–10128
Sewell AK, Jensen LT, Erickson JC, Palmiter RD, Winge DR (1995) Biochemistry 34:4740–4747
Palmiter RD (1995) Toxicol Appl Pharmacol 135:139–146
Irie Y, Keung WM (2001) Biochem Biophys Res Commun 282:416–420
Quaife CJ, Kelly EJ, Masters BA, Brinster RL, Palmiter RD (1998) Toxicol Appl Pharmacol 148:148–157
Penkowa M, Tio L, Giralt M, Quintana A, Molinero A, Atrian S, Vašák M, Hidalgo J (2006) J Neurosci Res 83:974–984
Hozumi I, Suzuki JS, Kanazawa H, Hara A, Saio M, Inuzuka T, Miyairi S, Naganuma A, Tohyama C (2008) Neurosci Lett 438:54–58
Yamada M, Hayashi S, Hozumi I, Inuzuka T, Tsuji S, Takahashi H (1996) Brain Res 735:257–264
Uchida Y, Gomi F, Masumizu T, Miura Y (2002) J Biol Chem 277:32353–32359
Lynes MA, Zaffuto K, Unfricht DW, Marusov G, Samson JS, Yin X (2006) Exp Biol Med (Maywood) 231:1548–1554
Chung RS, Penkowa M, Dittmann J, King CE, Bartlett C, Asmussen JW, Hidalgo J, Carrasco J, Leung YK, Walker AK, Fung SJ, Dunlop SA, Fitzgerald M, Beazley LD, Chuah MI, Vickers JC, West AK (2008) J Biol Chem 283:15349–15358
Klassen RB, Crenshaw K, Kozyraki R, Verroust PJ, Tio L, Atrian S, Allen PL, Hammond TG (2004) Am J Physiol Renal Physiol 287:F393–F403
Erickson JC, Hollopeter G, Thomas SA, Froelick GJ, Palmiter RD (1997) J Neurosci 17:1271–1281
Cole TB, Robbins CA, Wenzel HJ, Schwartzkroin PA, Palmiter RD (2000) Epilepsy Res 39:153–169
Aschner M, Cherian MG, Klaassen CD, Palmiter RD, Erickson JC, Bush AI (1997) Toxicol Appl Pharmacol 142:229–242
Knipp M, Meloni G, Roschitzki B, Vašák M (2005) Biochemistry 44:3159–3165
El Ghazi I, Martin BL, Armitage IM (2006) Exp Biol Med (Maywood) 231:1500–1506
Meloni G, Polanski T, Braun O, Vašák M (2009) Biochemistry 48:5700–5707
Assaf SY, Chung SH (1984) Nature 308:734–736
Palumaa P, Eriste E, Njunkova O, Pokras L, Jornvall H, Sillard R (2002) Biochemistry 41:6158–6163
Kang YJ (1999) Proc Soc Exp Biol Med 222:263–273
Liu J, Liu Y, Hartley D, Klaassen CD, Shehin-Johnson SE, Lucas A, Cohen SD (1999) J Pharmacol Exp Ther 289:580–586
Carrasco J, Penkowa M, Giralt M, Camats J, Molinero A, Campbell IL, Palmiter RD, Hidalgo J (2003) Neurobiol Dis 13:22–36
Chen Y, Irie Y, Keung WM, Maret W (2002) Biochemistry 41:8360–8367
Durand J, Meloni G, Talmard C, Vašák M, Faller P (2010) Metallomics 2:741–744
Lee SJ, Koh JY (2010) Mol Brain 3:30
Garrett SH, Sens MA, Shukla D, Nestor S, Somji S, Todd JH, Sens DA (1999) Prostate 41:196–202
Sens MA, Somji S, Garrett SH, Beall CL, Sens DA (2001) Am J Pathol 159:21–26
Tanji K, Irie Y, Uchida Y, Mori F, Satoh K, Mizushima Y, Wakabayashi K (2003) Brain Res 976:125–129
Dutta R, Sens DA, Somji S, Sens MA, Garrett SH (2002) Prostate 52:89–97
Karotki AV, Vašák M (2009) J Biol Inorg Chem 14:1129–1138
Bogumil R, Faller P, Binz PA, Vašák M, Charnock JM, Garner CD (1998) Eur J Biochem 255:172–177
Faller P, Vašák M (1997) Biochemistry 36:13341–13348
Hasler DW, Faller P, Vašák M (1998) Biochemistry 37:14966–14973
Faller P, Hasler DW, Zerbe O, Klauser S, Winge DR, Vašák M (1999) Biochemistry 38:10158–10167
Hagen KS, Stephan DW, Holm RH (1982) Inorg Chem 21:3928–3936
Öz G, Zangger K, Armitage IM (2001) Biochemistry 40:11433–11441
Wang H, Zhang Q, Cai B, Li H, Sze KH, Huang ZX, Wu HM, Sun H (2006) FEBS Lett 580:795–800
Hasler DW, Jensen LT, Zerbe O, Winge DR, Vašák M (2000) Biochemistry 39:14567–14575
Ni FY, Cai B, Ding ZC, Zheng F, Zhang MJ, Wu HM, Sun HZ, Huang ZX (2007) Proteins 68:255–266
Romero-Isart N, Oliva B, Vašák M (2010) J Mol Model 16:387–394
Romero-Isart N, Jensen LT, Zerbe O, Winge DR, Vašák M (2002) J Biol Chem 277:37023–37028
Ding ZC, Ni FY, Huang ZX (2010) FEBS J 277:2912–2920
Bruinink A, Faller P, Sidler C, Bogumil R, Vašák M (1998) Chem Biol Interact 115:167–174
Erickson JC, Sewell AK, Jensen LT, Winge DR, Palmiter RD (1994) Brain Res 649:297–304
Vašák M, Meloni G (2009) In: Sigel A, Sigel H, Sigel RKO (eds) Metallothioneins and related chelators. Royal Society of Chemistry, Cambridge, pp 319–351
Faller P (2010) FEBS J 277:2921–2930
Roschitzki B, Vašák M (2002) J Biol Inorg Chem 7:611–616
Jensen LT, Peltier JM, Winge DR (1998) J Biol Inorg Chem 3:627–631
Pountney DL, Schauwecker I, Zarn J, Vašák M (1994) Biochemistry 33:9699–9705
Roschitzki B, Vašák M (2003) Biochemistry 42:9822–9828
Gaggelli E, Kozlowski H, Valensin D, Valensin G (2006) Chem Rev 106:1995–2044
Barnham KJ, Masters CL, Bush AI (2004) Nat Rev Drug Discov 3:205–214
Sogawa CA, Asanuma M, Sogawa N, Miyazaki I, Nakanishi T, Furuta H, Ogawa N (2001) Acta Med Okayama 55:1–9
Kawashima T, Doh-ura K, Torisu M, Uchida Y, Furuta A, Iwaki T (2000) Dement Geriatr Cogn Disord 11:251–262
Hozumi I, Asanuma M, Yamada M, Uchida Y (2004) J Health Sci 50:323–331
Barnham KJ, Cappai R, Beyreuther K, Masters CL, Hill AF (2006) Trends Biochem Sci 31:465–472
Halliwell B, Gutteridge JM (1984) Biochem J 219:1–14
Meloni G, Faller P, Vašák M (2007) J Biol Chem 282:16068–16078
Meloni G, Sonois V, Delaine T, Guilloreau L, Gillet A, Teissie J, Faller P, Vašák M (2008) Nat Chem Biol 4:366–372
Chung RS, Howells C, Eaton ED, Shabala L, Zovo K, Palumaa P, Sillard R, Woodhouse A, Bennett WR, Ray S, Vickers JC, West AK (2010) PLoS One 5:e12030
Adlard PA, West AK, Vickers JC (1998) Neurobiol Dis 5:349–356
Richarz AN, Bratter P (2002) Anal Bioanal Chem 372:412–417
Zambenedetti P, Giordano R, Zatta P (1998) J Chem Neuroanat 15:21–26
Carrasco J, Adlard P, Cotman C, Quintana A, Penkowa M, Xu F, Van Nostrand WE, Hidalgo J (2006) Neuroscience 143:911–922
Adlard PA, Bush AI (2006) J Alzheimers Dis 10:145–163
Dawson TM, Dawson VL (2003) Science 302:819–822
Paik SR, Shin HJ, Lee JH (2000) Arch Biochem Biophys 378:269–277
Binolfi A, Lamberto GR, Duran R, Quintanar L, Bertoncini CW, Souza JM, Cervenansky C, Zweckstetter M, Griesinger C, Fernandez CO (2008) J Am Chem Soc 130:11801–11812
Drew SC, Leong SL, Pham CL, Tew DJ, Masters CL, Miles LA, Cappai R, Barnham KJ (2008) J Am Chem Soc 130:7766–7773
Aguzzi A, Heikenwalder M (2006) Nat Rev Microbiol 4:765–775
Prusiner SB (1991) Science 252:1515–1522
Meloni G, Vašák M (2011) Free Radic Biol Med 50:1471–1479
Liang L, Fu K, Lee DK, Sobieski RJ, Dalton T, Andrews GK (1996) Mol Reprod Dev 43:25–37
Schlake T, Boehm T (2001) Mech Dev 109:419–422
Meloni G, Zovo K, Kazantseva J, Palumaa P, Vašák M (2006) J Biol Chem 281:14588–14595
Cai B, Zheng Q, Huang ZX (2005) Protein J 24:327–336
Tio L, Villarreal L, Atrian S, Capdevila M (2004) J Biol Chem 279:24403–24413
Capdevila M, Domenech J, Pagani A, Tio L, Villarreal L, Atrian S (2005) Angew Chem Int Ed 44:4618–4622
Tio L, Villarreal L, Atrian S, Capdevila M (2006) Exp Biol Med (Maywood) 231:1522–1527
Prange A, Profrock D (2005) Anal Bioanal Chem 383:372–389
Mounicou S, Ouerdane L, L’Azou B, Passagne I, Ohayon-Courtes C, Szpunar J, Lobinski R (2010) Anal Chem 82:6947–6957
Acknowledgments
Parts of the work described in this review have been funded over the years by the Swiss National Science Foundation (M.V.). G.M. is a Marie Curie International Outgoing Fellow (European Commission, grant no. 252961).
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of a JBIC special issue on metallothioneins.
Rights and permissions
About this article
Cite this article
Vašák, M., Meloni, G. Chemistry and biology of mammalian metallothioneins. J Biol Inorg Chem 16, 1067–1078 (2011). https://doi.org/10.1007/s00775-011-0799-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-011-0799-2