
Abstract
DNA-binding proteins from starved cells (Dps) differ in the number and position of charged residues along the “ferritin-like” pores that are used by iron to reach the ferroxidase center and the protein cavity. These differences are shown to affect significantly the electrostatic potential at the pores, which determines the extent of cooperativity in the iron uptake kinetics and thereby the mass distribution of the ferric hydroxide micelles inside the protein cavity. These conclusions are of biotechnological value in the preparation of protein-enclosed nanomaterials and are expected to apply also to ferritins. They were reached after characterization of the Dps from Listeria innocua, Helicobacter pylori, Thermosynechococcus elongatus, Escherichia coli, and Mycobacterium smegmatis. The characterization comprised the calculation of the electrostatic potential at the pores, determination of the iron uptake kinetics in the presence of molecular oxygen or hydrogen peroxide, and analysis of the proteins by means of the sedimentation velocity after iron incorporation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Different structural elements take part in the iron oxidation/incorporation process, which endows the members of the ferritin superfamily—ferritins proper, bacterioferritins, and DNA-binding proteins from starved cells (Dps)—with iron detoxification/storage properties. Two such elements were identified in the ferritin X-ray crystal structure, namely, the protein cavity, where iron is incorporated and stored as ferric hydroxide micelles [1, 2], and the ferroxidase center, the site of Fe(II) oxidation [3]. In these studies, the hydrophilic, negatively charged pores formed along the threefold symmetry axes were suggested to provide the route taken by Fe(II) to reach the ferroxidase center. This contention was later proven to be true by means of spectroscopic experiments [4] and theoretical studies that entailed the calculation of the electrostatic potential at the pores [5, 6]. As expected, the pores have negative electrostatic potentials, with a major contribution by the negatively charged amino acids at the entrance of the pores, whereas the regions of positive potential surrounding the pores create an electrostatic field directed toward the interior cavity [5, 6].
Dps, even though their assembly (12-mers with 23 symmetry) differs relative to the characteristic ferritin one (24-mers with 432 symmetry), possess a ferroxidase center and a shell-like architecture traversed by hydrophilic, negatively charged pores (the so-called ferritin-like pores) at the threefold symmetry axes [7]. The participation of these pores in the iron uptake process was the subject of a recent study on Listeria innocua Dps (Li-Dps) which compared the behavior of the native protein with that of site-specific mutants lacking one or more of the negatively charged residues at the pores [8]. The corresponding decrease in the negative electrostatic potential was shown to affect the iron oxidation/incorporation kinetics, which in turn governs the mass distribution of the iron micelles in the protein cavity. Thus, the intense electrostatic gradient of the native protein results in marked kinetic cooperativity, which manifests itself in sigmoidal iron uptake progress curves, and hence favors formation of apo-Dps and fully iron loaded Dps molecules. The weakening of the electrostatic gradient in the site-specific mutants leads to quasi-hyperbolic progress curves, i.e., to lower cooperativity during iron uptake, and consequently to a rather homogeneous distribution of iron micelles in the Dps molecules [8].
The existence of a correlation between the number of negative charges along the ferritin-like pores, cooperativity in the iron uptake process, and mass distribution of the protein-enclosed iron micelles is of interest not only per se, since it likely applies to ferritins as well, but also from a biotechnological viewpoint. It can be exploited for the production of protein-enclosed nanominerals with homogeneous and predefined iron uptake properties, a difficult task to achieve otherwise [9–14]. Surprisingly, in the other members of the Dps family the iron uptake process has not been characterized in terms of kinetic cooperativity and the distribution of iron micelles, possibly because ferritins and bacterioferritins are the major players in the storage of bacterial iron [15–17].
Examination of the ferritin-like pores in the X-ray structures of Dps [18–24] reavels significant differences in the number and position of charged residues that, on the basis of the behavior of Li-Dps, are likely to result in markedly different iron uptake properties. In particular, two and three aspartic acid residues respectively line the pores of Escherichia coli Dps (Ec-Dps) and Li-Dps (D142 and D146 in Ec-Dps and D121, D126, and D130 in Li-Dps); two aspartic acid residues and one glutamic acid residue (E128, D129, and D136B) coat those of Mycobacterium smegmatis Dps (Ms-Dps), and positively charged residues are also located along the pores of Thermosynechococcus elongatus Dps (Te-Dps) and Helicobacter pylori Dps (Hp-Dps), namely, D130 and R134 in Te-Dps and E114, K119, D126, and D127 in Hp-Dps.
In this study, these five Dps were used for the calculation of the negative electrostatic potential at the ferritin-like pores and for the determination of the iron uptake kinetics and mass distribution of the iron micelles incorporated upon metal oxidation with O2 or H2O2. It may be recalled that among the selected Dps, Ec-Dps and Ms-Dps form large Dps-DNA complexes at neutral pH values owing to the interaction of their positively charged N- or C-termini with the negatively charged DNA backbone [25]. In the absence of this macromolecular partner, Ec-Dps and Ms-Dps tend to self-associate and precipitate out of solution unless their surface charges are shielded by the presence of 150–200 mM NaCl [23, 26].
As expected, on the basis of the considerations exposed, the variations in charge distribution that distinguish the selected Dps influence the iron uptake process markedly. However, the detailed effects related to the use of O2 or H2O2 as an iron oxidant and of different ionic strengths were difficult to anticipate because of the complexity of the iron uptake process and because of the disparate influence of the competing iron oxidation reaction in solution.
Materials and methods
Protein expression and purification
Ec-Dps, Li-Dps, Hp-Dps, Te-Dps, and Ms-Dps were obtained and purified as described previously [20, 23, 26–30]. The purified proteins typically contain less than five to six iron atoms per dodecamer.
Protein concentrations were determined spectrophotometrically at 280 nm on the basis of the molar extinction coefficients (on a dodecamer basis, in milliliters per milligram per centimeter), ε: 0.83 (M = 221 kDa) for Ec-Dps, 0.94 (M = 215 kDa) for Li-Dps, 1.0 (M = 202 kDa) for Hp-Dps, 1.11 (M = 215 kDa) for Te-Dps, and 1.10 (M = 243 kDa) for Ms-Dps.
Iron oxidation and incorporation
The Fe(II) oxidation/incorporation kinetics caused by O2 and H2O2 in the presence of the Dps under study was followed at 450 nm with a Hewlett-Packard (Palo Alto, CA, USA) diode-array spectrophotometer at a constant temperature of 25 °C. All the traces were analyzed with Origin 8 (OriginLab, Northampton, MA, USA).
Ferrous sulfate was used as the Fe(II) source. Stock solutions (25 mM) were prepared freshly in Thunberg tubes prior to the experiments and used within a few hours of their preparation. When O2 was used as the Fe(II) oxidant, 250 Fe(II) atoms per dodecamer were added in a single addition to 2 μM apo-Dps solutions kept under constant stirring in air; when H2O2 was used (at a 1:2 molar ratio with respect to iron), the metal was added in five steps of 50 Fe(II) per dodecamer to 1 μM apo-Dps solutions. The Dps that do not bind DNA were in 100 mM 3-(N-morpholino)propanesulfonic acid (MOPS)–NaOH buffer pH 7.0, and those which bind DNA were in 50 mM MOPS–NaOH containing 200 mM NaCl. At the end of the experiments, to avoid formation of ferric hydroxide aggregates and their precipitation, sodium citrate was added in a 1:1 molar ratio relative to iron. As a control, the rate of Fe(II) autooxidation was measured in parallel. The ferric hydroxide aggregates formed during Fe(II) autooxidation under the conditions used tend to precipitate out of solution as indicated by the decrease in absorbance during the later stages of the reaction.
Analytical ultracentrifugation
Sedimentation velocity experiments were carried out with a Beckman Optima XL-I analytical ultracentrifuge using absorbance optics. Experiments were conducted at 30,000 rpm and 20 °C at the Dps concentrations (1.0 or 2.0 μM) and in the buffers used in the iron oxidation/incorporation kinetic experiments. Radial absorbance scans were obtained in a continuous scan mode at 280 and 350 nm at a spacing of 0.003 cm; three scans were averaged. Sedimentation coefficients were calculated using the program Sedfit (provided by P. Schuck, National Institutes of Health) and were reduced to water and 20 °C (s 20,w) using standard procedures. The buffer density and viscosity were calculated by the program Sednterp. The particle size distribution was calculated by converting the sedimentation coefficient distribution into size distributions, and taking into account the density and thickness of the protein shell (1.35 g/cm3 and 2 nm, respectively) and the density of the ferric core in the form of maghemite (3.8 g/cm3) [25].
Calculation of electrostatic potentials
The electrostatic potentials were calculated from the X-ray crystal structures of the chosen Dps (Table 1). The amino acids that are not visible in the X-ray structures were added by molecular modeling as reported in the same table. The N-terminal regions of the Dps monomers were built using the program SYBYL 6.0 (http://www.tripos.com), whereas the 19 residues missing at the C-terminus of Ms-Dps were modeled with the I-Tasser server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) [31]. The structure of the monomer was used as a reference for the orientation of the fragment modeled by I-Tasser that has been extended toward solvent.
The calculations were performed through the program APBS [32], which solves the Poisson–Boltzmann equation by a finite difference method using the charges of the AMBER force field [33].
To calculate the electrostatic potential, the protein was placed into a cubic grid with a 110–120-Å side, depending on the dodecamer size. The grid spacing was 0.5 Å for a total of about 12 million potential values calculated. The potential was calculated for each channel, overlapping the axis passing through the pore on the x-axis of the grid. To ensure the correct orientation, a program for the roto-translation of the dodecamer was designed specifically. The axes chosen for the calculation cross the four iron-entry pores perpendicularly and converge toward the center of the dodecamer. The zero value of the axis corresponds to the geometric center calculated between the residues that form the inner edge of the pore. The axis of the electrostatic potential extends from −10 Å (inside the pore toward the center of the dodecamer) to +15 Å (over the pore toward the outer surface of the dodecamer). The potential values are expressed in units of kT/e (where k is the Boltzmann constant, T = 298 K, e is the electronic charge, and 1 kT/e = 0.0257 V).
In the calculation of electrostatic potentials, the APBS program assumes the same value of ionic strength both inside and outside the dodecamer cavity. The ionic strength within the Dps cavity is not known and it is likewise unknown how it is influenced by the ionic selectivity of the pores. For this reason, to highlight solely the contribution of the protein structure, we chose to compare the electrostatic potentials calculated at zero ionic strength.
The modelled monomer chains are indicated by capital letters. The addition of the indicated amino acids to the respective monomers does not affect the electrostatic potential of the dodecamer.
Results
Iron oxidation/uptake in the presence of molecular oxygen
The electrostatic potential at the ferritin-like pores will be illustrated prior to the iron incorporation data to underscore its relevance in determining the kinetic features of the uptake process and the consequent distribution of the protein-enclosed iron micelles.
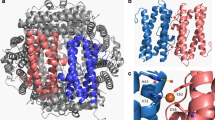
Figure 1 shows the electrostatic surface of Li-Dps, Te-Dps, and Hp-Dps, including a blowup of the pores with the charged residues highlighted, and the electrostatic potential calculated along the pores at zero ionic strength (see “Materials and methods” for details). Li-Dps is characterized by a strong negative potential due mainly to the three aspartic acid residues (namely, D121, D126, and D130) that line the pores. The electrostatic potential changes from about −70 kT/e at the external surface to about −270 kT/e within a 2 Å distance toward the center of the molecule. In Te-Dps, the electrostatic potential is significantly less marked owing to the presence along the pores of both negative and positive residues, D130 and R134, respectively. Thus, it changes from −25 to −175 kT/e over a similar distance when moving from the external surface of the molecule toward its center. A different situation applies to Hp-Dps given the unusually high number of positively charged residues on the molecular surface and the presence of negative (E114, D126, and D127) and positive (K119) residues on the rather wide pores. The resulting electrostatic potential is shallow and changes from −25 kT/e at the external surface to −175 kT/e over a distance of about 20 Å toward the internal cavity.

DNA-binding proteins from starved cells (Dps) from Listeria innocua (Li-Dps) (a), Thermosynechococcus elongatus (Te-Dps) (b), and Helicobacter pylori (Hp-Dps) (c): protein surface colored according to charge, “ferritin-like” pores, and electrostatic potential at the pores. Red negatively charged residues, blue positively charged residues, white uncharged residues. Highlighted residues in Li-Dps (a) D121, D126, and D130; in Te-Dps (b) D130 and R134; in Hp-Dps (c) E114, K119, D126, and D127. The figure was created with the program PyMOL (version 0.99) [34]. The electrostatic potential is measured as a function of distance (from −10 to +15 Å) from the geometric center of the pore and is expressed in kT/e
These electrostatic potential differences are reflected in the kinetics of iron uptake in the presence of O2 as expected from the work of Bellapadrona et al. [8]. Iron uptake was followed under the experimental conditions used in this work, namely, 25 °C in 100 mM MOPS, pH 7.0 (I = 28 mM), after a single addition of 250 Fe(II) atoms per dodecamer so as to reach half the Dps iron storage capacity [27]. The rationale for adding iron in one aliquot rather than several aliquots resides in the very nature of the uptake process. When the amount of iron added exceeds by far the binding capacity of the ferroxidase center (24 Fe(II) atoms per dodecamer), the contribution of the electrostatic gradient is the largest since iron oxidation on the growing ferric micelle becomes relevant [8].
The steep negative potentials of Li-Dps and Te-Dps give rise to sigmoidal progress curves, whereas the shallow potential of Hp-Dps yields quasi-hyperbolic iron uptake kinetics (Fig. 2). The sigmoidal progress curves point to a complex kinetic landscape and are typical of crystal formation where a slow nucleation step is followed by fast growth of the crystal. In mechanistic terms, the corresponding increase in rate is often referred to as kinetic cooperativity [8]. Thus, kinetic cooperativity, evidenced by the derivative of the progress curve (Fig. 2, insets), is marked in Li-Dps and Te-Dps, but almost zero in Hp-Dps. Consequently, the size distribution of the protein-enclosed iron micelles assessed in sedimentation velocity experiments differs. Hence, the patterns pertaining to Li-Dps and Te-Dps reflect a highly cooperative process which favors formation of two distinct populations, namely, apoprotein (10 S) and fully mineralized protein (19.5 S). In contrast, iron-incorporated Hp-Dps sediments as a single fairly homogeneous peak whose velocity (14.3 S) is intermediate between the velocities of the apoprotein and the fully loaded protein, in accord with the decreased cooperativity of the uptake process which favors formation of partially filled protein molecules (Fig. 2). It is worth noting that the protein-catalyzed rates are faster than the rate of iron autooxidation such that all the iron added is incorporated in the protein cavity.

Iron oxidation/uptake of 250 Fe(II) atoms per dodecamer by Li-Dps (a), Te-Dps (b), and Hp-Dps (c) in the presence of O2 as an oxidant. Left Incorporation kinetics in 100 mM 3-(N-morpholino)propanesulfonic acid (MOPS) pH 7.0 (I = 28 mM); the red curve represents the autooxidation of Fe(II) in absence of protein. The inset shows the first derivative of the process (the red interpolated curve was obtained with the program Origin 8). Right Sedimentation profile of the incorporated samples
To confirm the relevance of kinetic cooperativity in determining the size distribution of the ferric cores, 250 Fe(II) atoms per dodecamer were added in steps of 50 atoms to Li-Dps. The kinetics of iron uptake is hyperbolic (Fig. 3). As a result, a situation resembling that of Hp-Dps is realized: no apoprotein peak is present at the end of the reaction, and the mass distribution of the ferric cores is significantly wider than in the case of a single-step addition (cf. Fig. 2a).

Iron oxidation/uptake of 250 Fe(II) atoms per dodecamer by Li-Dps added in 50 atoms per step in the presence of O2 as an oxidant. a Kinetics of iron oxidation/uptake by Li-Dps in 100 mM MOPS at pH 7.0 (I = 28 mM). b Sedimentation profile at the end of the reaction
Ec-Dps and Ms-Dps bind DNA at neutral pH values, at variance with Li-Dps, Te-Dps, and Hp-Dps, and tend to self-aggregate in the absence of this macromolecular partner at ionic strengths below 150–200 mM [23, 26]. Figure 4 depicts the respective electrostatic surface, ferritin-like pores, and electrostatic potential at the pores. In Ec-Dps, the D142 and D146 residues lining the pores give the major contribution to the potential, which changes from −75 to −175 kT/e over about 1 Å in going from the external surface to the protein cavity. The pattern therefore resembles that of Te-Dps (see Fig. 2). In Ms-Dps, the electrostatic potential, due primarily to the E128, D129, and D136 residues along the pores, changes in a stepwise fashion from −30 to −270 kT/e over a distance of roughly 20 Å.

Escherichia coli Dps (Ec-Dps) (a) and Mycobacterium smegmatis Dps (Ms-Dps) (b): protein surface colored according to charge, ferritin-like pores, and electrostatic potential at the pores. Red negatively charged residues, blue positively charged residues, white uncharged residues. Highlighted residues: in Ec-Dps (a) D142 and D146; in Ms-Dps (b) E128, D129 and D136. The figure was created with PyMOL. The electrostatic potential is measured as a function of distance (from −10 to +15 Å) from the geometric center of the pore and is expressed in kT/e. In one of the pores of Ms-Dps, three glutamine residues have a different conformation than in the other pores; this gives rise to the slightly different potential indicated by the red line
The kinetics of iron uptake by Ec-Dps and Ms-Dps was followed in 50 mM MOPS buffer containing 200 mM NaCl (I = 214 mM). Figure 5 shows that salt decreases the iron oxidation rate such that the rate of autooxidation becomes faster than the protein-catalyzed oxidation (see Fig. 2). As a result, despite the sigmoidal character of the iron uptake kinetics, a significant part of iron is oxidized outside the protein. Accordingly, the sedimentation pattern of iron-incorporated Ec-Dps displays mostly apoprotein (10 S) and little fully loaded protein (18.1 S) and iron-incorporated Ms-Dps shows small amounts of peaks/shoulders at 18, 26, and 44.5 S. These can be attributed to iron hydroxide aggregates that are maintained in solution by sodium citrate and by the relatively high ionic strength.

Iron oxidation/uptake of 250 Fe(II) atoms per dodecamer by Ec-Dps (a) and Ms-Dps (b) in the presence of O2 as an oxidant. Left Incorporation kinetics in 50 mM MOPS pH 7.0 containing 200 mM NaCl (I = 214 mM); The red curve represents the autooxidation of Fe(II) in the absence of protein. Right Sedimentation profile of the incorporated samples
The behavior of Ec-Dps and Ms-Dps just described prompted a systematic study of the effect of ionic strength on the iron uptake kinetics of the Dps that do not bind DNA. Firstly, Li-Dps was studied over the ionic strength range from 5.6 to 214 mM. The higher ionic strengths, namely, 85.6 and 214 mM, were obtained by addition of NaCl at increasing concentrations. At the same time, the concentration of MOPS was likewise increased to enhance its buffering capacity. Figure 6 shows clearly that the increase in ionic strength hardly affects the sigmoidal character of the progress curve. However, it slows down the protein-catalyzed oxidation reaction significantly more than autooxidation, such that the two rates are very similar at the highest ionic strength studied. The distribution of the iron micelles incorporated in the Li-Dps cavity varies accordingly. At the lower ionic strengths investigated (5.6 and 28 mM, corresponding to 20 and 100 mM MOPS at pH 7.0, respectively), apoprotein (10 S) and fully loaded protein (19 S) are both present in substantial amounts. At the higher ionic strengths, apo-Dps is predominant, indicating that iron is oxidized preferentially outside the protein cavity. Thus, the sedimentation pattern at I = 85.6 mM (20 mM MOPS at pH 7.0 containing 80 mM NaCl) shows partially loaded protein molecules (s 20,w ~ 17 S) and significant amounts of soluble ferric hydroxide aggregates (s 20,w > 20 S). At I = 214 mM (50 mM MOPS containing 200 mM NaCl), ferric hydroxide aggregation outside the protein increases further, such that the ferric aggregates become larger and insoluble as indicated by the decrease in concentration of the soluble protein.

Effect of ionic strength on the kinetics of Fe(II) oxidation/uptake by Li-Dps and on the distribution of iron micelles in the protein cavity in the presence of O2. Top Iron oxidation/uptake kinetics upon addition of 250 Fe(II) atoms per dodecamer in MOPS–NaOH buffer containing NaCl at pH 7.0 of the indicated ionic strength; the red curve represents the autooxidation of Fe(II) in the absence of protein. In view of the sigmoidal shape of the progress curves, the ionic strength effect on the overall rate of the process was assessed by comparing the t 1/2 values calculated from the final absorbance value at 450 nm, corresponding to 99% oxidation of Fe(II). From left to right the t 1/2 values are 92, 222, 480 and 2,240 min. Bottom Sedimentation profile of the incorporated samples
To establish the generality of these results, i.e., that a decrease in ionic strength favors iron incorporation, Te-Dps and Hp-Dps were assayed at 5.6 mM ionic strength (20 mM MOPS at pH 7.0). The sedimentation patterns (Fig. 7) show distinct differences relative to those obtained at I = 28 mM (100 mM MOPS) depicted in Fig. 2. The iron micelles are distributed in a considerably more homogeneous fashion. Thus, in Te-Dps there is only a small shoulder attributable to the apoprotein (s 20,w ~ 11.8 S), whereas most molecules give rise to a homogeneous peak at 16.6 S. In Hp-Dps, only a single peak centered at approximately 15.5 S, pertaining to iron-containing molecules, is observed. This assignment was confirmed in gel filtration experiments (not shown).

Sedimentation profiles of Te-Dps (a) and Hp-Dps (b) after oxidation/uptake of 250 Fe(II) atoms per dodecamer in the presence of O2 in 20 mM MOPS–NaOH buffer at pH 7.0 (I = 5.6 mM)
The sedimentation velocity data obtained and the estimate of the size of the iron core formed in the various Dps under the ionic strength conditions analyzed are compiled in Table 2.
Iron oxidation by hydrogen peroxide
Hydrogen peroxide is a more efficient Fe(II) oxidant than oxygen for the vast majority of Dps and the rate of Fe(II) oxidation is about 100-fold higher than in the presence of molecular oxygen [7, 27]. In the presence of H2O2, Fe(II) is added in small amounts to achieve effective iron incorporation in the Dps cavity and avoid Fe(III) precipitation outside the protein. Therefore, the experiments were carried out in 50 mM MOPS buffer (I = 14 mM) at pH 7.0 and entailed five successive additions of 50 Fe(II) atoms per dodecamer.
In Li-Dps, Te-Dps, and Hp-Dps the progress curves are hyperbolic and iron is incorporated homogeneously in all the protein molecules as shown by the sedimentation velocity peaks centered at 12.8, 12.5, and 13 S, respectively. In the case of Te-Dps, there is also a small peak at 20 S due to the formation of soluble Fe(III) aggregates outside the protein shell (Fig. 8).

Iron oxidation/uptake upon successive additions of 50 Fe(II) atoms per dodecamer in the presence of H2O2 as an oxidant. a Kinetics of iron oxidation/uptake by Li-Dps in 100 mM MOPS at pH 7.0 (I = 28 mM); the curve at the bottom represents the autooxidation of Fe(II) in the absence of protein. Sedimentation profiles after the oxidation/uptake reaction of Li-Dps (b), Te-Dps (c), and Hp-Dps (d)
Ec-Dps and Ms-Dps, the two DNA-binding proteins investigated, were analyzed in the same 50 mM MOPS buffer containing 200 mM NaCl (I = 214 mM). Massive precipitation of ferric hydroxide aggregates takes place and very little iron is incorporated. Thereafter, Li-Dps was analyzed at this ionic strength. The iron micelles grown in the protein cavity are distributed in a considerably more heterogeneous fashion than at I = 14 mM. To facilitate comparison with iron uptake in the presence of molecular oxygen, the sedimentation velocity data have been included in Table 2.
Discussion
This survey of the iron uptake process by Dps differing in number and position of charged residues at the ferritin-like pores illustrates the functional effects of such differences under the various conditions used typically in iron oxidation/uptake experiments. These effects have never been analyzed systematically even though they could be expected to be large on theoretical grounds as they influence the negative electrostatic potential at the pores and thereby the force driving iron toward the catalytic centers and the protein cavity [5, 6].
The sedimentation velocity data summarized in Table 2 show that the amount of iron taken up by a given Dps and the distribution of the metal within the protein population depend on the outcome of the competition between concomitant phenomena [namely, Fe(II) autooxidation, iron oxidation/uptake by the protein, Fe(II) oxidation outside the protein shell and formation of Fe(III) aggregates] whose contribution varies depending on the experimental conditions used [nature of the Fe(II) oxidant and ionic strength].
The consequences of using O2 or H2O2 as an iron oxidant will be discussed first. Molecular oxygen is characterized by slower Fe(II) oxidation rates with respect to H2O2 such that relatively large amounts of iron [up to 300 Fe(II) atoms per dodecamer] can be added to the protein solutions at neutral pH without precipitation of ferric hydroxide aggregates outside the protein shell. Iron incorporation is cooperative and favors full loading of the protein cavity in only part of the molecules (Fig. 2). The more efficient H2O2, on the other hand, requires that iron be added in small amounts, e.g., 50 Fe(II) atoms per dodecamer (Fig. 8a), to minimize iron oxidation outside the protein shell. In turn, these conditions result in the absence of cooperativity in iron uptake and in a homogenous distribution of iron micelles in all the Dps molecules (Fig. 8b–d). These observations are of value from a biotechnological perspective, since the dependence of the iron uptake kinetics and thereby of the size and homogeneity of the protein-enclosed iron micelles on the nature of the oxidant can be exploited in the preparation of nanoparticles with desired characteristics. On the other hand, their physiological relevance is difficult to predict. Thus, the iron oxidant may differ depending on the environment or growth conditions (e.g., pathogenic bacteria will prefer H2O2 during host infection when its production increases considerably) as well as iron availability.
The interplay between O2 as an iron oxidant and ionic strength was difficult to anticipate. The most extensive study was carried out over the range I = 5.6–214 mM using Li-Dps as a model protein. The Fe(II) oxidation rate is decreased, in particular in the presence of protein, and the solubility of ferric hydroxide is increased at I ≥ 30 mM. As a result, with increase in ionic strength, Fe(II) autooxidation competes more effectively with the protein-catalyzed reaction, whose efficiency decreases. However, the Fe(III) aggregates formed outside the protein tend to remain in solution even under the action of the centrifugal force (Fig. 6).
In conclusion, the complex pattern that links iron uptake by Dps to the hitherto neglected charge distribution at the ferritin-like pores, to the nature of the oxidant, and to ionic strength leads to an unsuspected functional versatility of clear physiological and biotechnological value also because a similar linkage pattern most likely applies to ferritins.
References
Banyard SH, Stammers DK, Harrison PM (1978) Nature 271:282–284
Ford GC, Harrison PM, Rice DW, Smith JM, Treffry A, White JL, Yariv J (1984) Philos Trans R Soc Lond B Biol Sci 304:551–565
Lawson DM, Treffry A, Artymiuk PJ, Harrison PM, Yewdall SJ, Luzzago A, Cesareni G, Levi S, Arosio P (1989) FEBS Lett 254:207–210
Stefanini S, Desideri A, Vecchini P, Drakenberg T, Chiancone E (1989) Biochemistry 28:378–382
Douglas T, Ripoll DR (1998) Protein Sci 7:1083–1091
Takahashi T, Kuyucak S (2003) Biophys J 84:2256–2263
Chiancone E, Ceci P (2010) Biochim Biophys Acta Gen Subj 1800:798–805
Bellapadrona G, Stefanini S, Zamparelli C, Theil EC, Chiancone E (2009) J Biol Chem 284:19101
Parker MJ, Allen M, Ramsay B, Klem TM, Young MJ, Douglas T (2008) Chem Mater 20:1541–1547
Gu QF, Krauss G, Steurer W, Gramm F, Cervellino A (2008) Phys Rev Lett 100:045502
Elnathan R, Kantaev R, Patolsky F (2008) Nano Lett 8:3964–3972
Kasyutich O, Ilari A, Fiorillo A, Tatchev D, Hoell A, Ceci P (2010) J Am Chem Soc 132:3621–3627
Suzuki M, Abe M, Ueno T, Abe S, Goto T, Toda Y, Akita T, Yamada Y, Watanabe Y (2009) Chem Commun 4871–4873
Lee LA, Wang Q (2006) Nanomedicine 2:137–149
Harrison PM, Arosio P (1996) Biochim Biophys Acta 275:161–203
Carrondo MA (2003) EMBO J 22:1959–1968
Theil EC (2004) Annu Rev Nutr 24:327–343
Grant RA, Filman DJ, Finkel SE, Kolter R, Hogle JM (1998) Nat Struct Biol 5:294–303
Ilari A, Stefanini S, Chiancone E, Tsernoglou D (2000) Nat Struct Mol Biol 7:38–43
Franceschini S, Ceci P, Alaleona F, Chiancone E, Ilari A (2006) FEBS J 273:4913–4928. doi:10.1111/j.1742-4658.2006.05490.x
Roy S, Gupta S, Das S, Sekar K, Chatterji D, Vijayan M (2004) J Mol Biol 339:1103–1113
Ceci P, Ilari A, Falvo E, Giangiacomo L, Chiancone E (2005) J Biol Chem 280:34776–34785. doi:10.1074/jbc.M502343200
Ceci P, Ilari A, Falvo E, Chiancone E (2003) J Biol Chem 278:20319–20326. doi:10.1074/jbc.M302114200
Zanotti G, Papinutto E, Dundon WG, Battistutta R, Seveso M, Giudice GD, Rappuoli R, Montecucco C (2002) J Mol Biol 323:125–130
Chiancone E, Ceci P (2010) Front Biosci 15:122–131
Ceci P, Cellai S, Falvo E, Rivetti C, Rossi GL, Chiancone E (2004) Nucleic Acids Res 32:5935–5944. doi:10.1093/nar/gkh915
Zhao G, Ceci P, Ilari A, Giangiacomo L, Laue TM, Chiancone E, Chasteen ND (2002) J Biol Chem 277:27689–27696. doi:10.1074/jbc.M202094200
Ilari A, Latella MC, Ceci P, Ribacchi F, Su M, Giangiacomo L, Stefanini S, Chasteen ND, Chiancone E (2005) Biochemistry 44:5579–5587. doi:10.1021/bi050005e
Ceci P, Mangiarotti L, Rivetti C, Chiancone E (2007) Nucleic Acids Res 35:2247–2256. doi:10.1093/nar/gkm077
Ceci P, Chiancone E, Kasyutich O, Bellapadrona G, Castelli L, Fittipaldi M, Gatteschi D (2010) Chemistry 16:709–717
Zhang Y (2009) Prot Struct Funct Bioinform 77:100–113. doi:10.1002/prot.22588
Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Proc Natl Acad Sci USA 98:10037–10041
Ponder JW, Case DA (2003) Adv Prot Chem 66:27–85
DeLano WL (2002) The PyMOL molecular graphics system. DeLano Scientific, San Carlos
Acknowledgments
E.C. acknowledges the support of local grants from the Ministero dell’Università e Ricerca Scientifica (MIUR). F.O thanks Filas for a fellowship granted under the project “Caratterizzazione di principi attivi.”
Author information
Authors and Affiliations
Corresponding author
Additional information
P. Ceci and G. Di Cecca contributed equally to the work.
Rights and permissions
About this article
Cite this article
Ceci, P., Di Cecca, G., Falconi, M. et al. Effect of the charge distribution along the “ferritin-like” pores of the proteins from the Dps family on the iron incorporation process. J Biol Inorg Chem 16, 869–880 (2011). https://doi.org/10.1007/s00775-011-0784-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-011-0784-9