
Abstract
Our previous study indicated that vanadium compounds can block cell cycle progression at the G1/S phase in human hepatoma HepG2 cells via a highly activated extracellular signal-regulated protein kinase (ERK) signal. To explore their differential action on normal cells, we investigated the response of an immortalized hepatic cell line, L02 cells. The results demonstrated that a higher concentration of vanadium compounds was needed to inhibit L02 proliferation, which was associated with S and G2/M cell cycle arrest. In addition, in contrast to insignificant reactive oxygen species (ROS) generation in HepG2 cells, all of the vanadium compounds resulted significant increases in both O ·−2 and H2O2 levels in L02 cells. At the same time, ERK and c-Jun N-terminal kinase (JNK) as well as cell division control protein 2 homolog (Cdc2) were found to be highly phosphorylated, which could be counteracted with the antioxidant N-acetylcysteine (NAC). The current study also demonstrated that both the ERK and the JNK pathways contributed to the cell cycle arrest induced by vanadium compounds in L02 cells. More importantly, it was found that although NAC can ameliorate the cytotoxicity of vanadium compounds in L02 cells, it did not decrease their cytotoxicity in HepG2 cells. It thus shed light on the potential therapeutic applications of vanadium compounds with antioxidants as synergistic agents to reduce their toxicities in human normal cells without affecting their antitumor activities in cancer cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vanadium compounds have been considered as a new class of metal-based antitumor agents in recent decades [1]. However, it has also been reported that they have a potential for carcinogenetic action [2–6]. This presents a potentially significant safety concern relating to their application as antitumor agents, and so a deeper understanding of the mechanism of their action is necessary.
Two seemingly “universal” mechanisms have been proposed on the basis of the chemistry and biochemistry of vanadium compounds [1, 7]. One is based on the interconversion between V(IV) and V(V) in biological media [8, 9], and vanadium compounds are assumed to be reactive oxygen species (ROS)-generating agents. Thus, many researchers have attributed their anticancer and cytotoxic effects, as well as their carcinogenetic properties, to induced oxidative stress and free radical production [1, 10–12]. The other mechanism is based on the structural similarity of vanadate to phosphate, and so it assumes that vanadium compounds can exert their effects on signal transduction pathways as the transition-state analogs of the protein tyrosine phosphatases [7, 13]. However, the two mechanisms are somewhat ubiquitous and lack specificity, since ROS and phosphatases are well known to mediate signaling pathways involved in almost all cellular processes. How vanadium compounds integrate all of the induced signals into a specific biological effect or possibly a dominant one in various cell types is therefore still a mystery. For example, it was previously demonstrated that vanadate was able to induce [14], suppress [15], or enhance [16] apoptosis. Although different solid forms of pentavalent vanadate may be used in these studies, in cell culture medium, solutions derived from these different solid forms should be an indistinguishable mixture containing [H2VO4]−, [HVO4]2− and some oligovanadates [17]. In addition, since the treatment time used in all of the above studies to induce the biological effects of vanadate was over 24 h, the incubation period can be excluded from the factors that lead to their specific biological effects. So, it seems that vanadate may cause a cell-specific but contradictory result that underlies the “universal mechanisms.” Apparently, differences in the genetic backgrounds of the cell types, such as expression of tumor suppressors or oncogenes and redox states of various cells, may act as biological determinants that allow vanadium compounds to exert cell-specific effects. Therefore, to optimize therapeutic potential and minimize possible side effects, the specific conditions present in biological systems should be investigated. Moreover, any anticancer agent must show selectivity towards cancer rather than normal cells [18]. Therefore, to develop potential anticancer vanadium compounds, their effects on both cancer cells and their normal counterparts must be considered. In our previous study, we demonstrated that three well-known antidiabetic compounds, sodium metavanadate (NaVO3), bis(maltolato)-oxovanadium(IV) (VO(ma)2) [19, 20], and bis(acetylacetonato)-oxovanadium(IV) (VO(acac)2) [21], did not induce significant ROS generation at concentrations that caused cell cycle arrest in human hepatoma HepG2 cells. They blocked cell cycle progression at the G1/S phase via a highly activated extracellular signal-regulated protein kinase (ERK) signal [22]. In the present study, to clarify whether and how vanadium compounds exert differential actions in normal liver cells and cancer cells, and determine whether they cause potential damage to normal cells, the effects of vanadium compounds on human hepatoma HepG2 and hepatic L02 cells are compared, and the underlying mechanism is explored.
Materials and methods
Materials
Bis(maltolato)-oxovanadium(IV) (VO(ma)2) was a generous gift from Prof. Debbie C. Crans, Colorado State University, Fort Collins, CO, USA. Sodium metavanadate (NaVO3), bis(acetylacetonato)-oxovanadium(IV) (VO(acac)2), N-acetyl cysteine (NAC), 3,8-diamino-5-[3-(diethylmethylamino)propyl]-6-phenyl phenanthridinium diiodide [propidium iodide, (PI)], ribonuclease A (RNAse A), and 1,9-pyrazoloanthrone (SP600125) were purchased from Sigma–Aldrich (St. Louis, MO, USA). MEK inhibitor I was obtained from Calbiochem (Darmstadt, Germany). Fetal bovine serum (FBS), Minimum Essential Medium and RPMI 1640 medium were obtained from Gibco (Santa Clara, CA, USA). Dihydroethidium (HE) and 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) were obtained from Molecular Probes (Eugene, OR, USA). 3-(4,5-Dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) were from Amresco (Solon, OH, USA). Phospho-pRb (Ser 780), phospho-pRb (Ser 807/811), phospho-Akt (Ser 473), phospho-ERK (Thr 202/Tyr 204), ERK, phospho-JNK (Thr 183/Tyr 185), JNK, phospho-p38 (Thr 180/Tyr 182), p38, phospho-Cdc2 (Tyr 15), Cdc2, β-actin, GAPDH-specific antibodies and anti-rabbit antibodies conjugated to horseradish peroxidase were from Cell Signaling Technology (Beverly, MA, USA). Anti-Cyclin D1 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Amersham ECL Plus western blotting detection reagents were from GE Healthcare (Amersham, UK). All other reagents were of analytical grade.
Cell culture
Human hepatoma HepG2 cells were obtained from the Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China), and human normal liver L02 cells were purchased from the Chinese Academy of Sciences (Shanghai, China). HepG2 and L02 cells were cultured in Minimum Essential Medium and RPMI 1640 medium, respectively. Both media were supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, 10 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid and 1 mM sodium pyruvate. The cells were cultured in a humidified atmosphere of 5% CO2 at 37 °C.
Cell viability assays
For the cell viability measurements, 5,000 cells (in 200 μl medium supplemented with 10% FBS) were seeded into each well of 96-well microplates, grown for 24 h, then treated with or without vanadium compounds for 24, 48 or 72 h. Afterwards, the tetrazolium salt MTT was added to each well to a final concentration of 0.5 mg/ml, and the cells were cultured in the CO2 incubator for an additional 4 h. The formazan crystals were dissolved in 200 μl DMSO for 30 min at 37 °C. Then the absorbance was measured at 570 nm by a microplate reader (Multiskan Mk3, Thermo Labsystems, Vantaa, Finland). The cell viability was calculated by normalizing the absorbance to the corresponding control.
To avoid the effects of thiol-containing antioxidant compounds (e.g., N-acetyl cysteine) on reducing MTT tetrazolium salts, which are reported to potentially lead to false-positive results in the MTT assay [23, 24], cell counting with an improved Neubauer hemocytometer was applied. Briefly, HepG2 and L02 cells were seeded at 3 × 104 per well in 24-well flat-bottomed plates and incubated for 24 h. Cells were then exposed to the indicated agents for another 24 h. After that, the supernatants were removed and the adherent cells were harvested by trypsinization. Cell suspension of each treatment group was diluted to a fixed volume and introduced into the counting chamber of the hemocytometer. The total number of cells was determined under the microscope and compared to the untreated control (normalized to 100% viability).
Cell cycle analysis by flow cytometry
HepG2 and L02 cells were plated onto 3.5 cm Petri dishes and incubated at 37 °C for 24 h. After serum starvation for 24 h in media containing 0.1% FBS, the synchronized cells were treated with or without vanadium compounds in the media containing 10% FBS. At different time points, cells were washed twice with cold phosphate-buffered saline containing 1 mM ethylenediaminetetraacetic acid (PBS-EDTA). Both floating and adherent cells were collected by trypsinization and centrifugation at 1,000 rpm for 5 min. The cells were then fixed in 80% ethanol (−20 °C). Before analysis, the supernatant was removed by centrifugation at 1,700 rpm for 5 min. Cells were washed once in cold PBS-EDTA and resuspended in PBS-EDTA containing 40 μg/ml PI and 100 μg/ml RNAse A. The cell cycle distribution was analyzed by flow cytometry (FACSCalibur, BD Biosciences, San Jose, CA, USA) using CellQuest Pro software (BD Biosciences).
Western blotting
HepG2 and L02 cells were incubated in six-well flat-bottomed plates or 3.5 cm dishes for 24 h. After synchronization by serum deprivation as described above, the cells were exposed to different agents at indicated intervals in the presence of 10% FBS. Then cells were harvested in lysis buffer and subjected to sonication. If necessary, a 20-min centrifugation at 10,000 rpm was performed to remove the precipitated fragments. The protein concentration of the supernatant was measured by Bradford protein assay using bovine serum albumin (BSA) as a standard. Equal amounts of total proteins were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE, Bio Rad Laboratories, Inc.). Gels were then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore Corperation). Transblotted PVDF membranes were blocked with 5% BSA in Tris-buffered saline (pH 7.6) containing 0.1% Tween 20 (TBST) for 1 h and then incubated overnight with the indicated primary antibody in 1% BSA-TBST at 4 °C. After being washed thrice with TBST for a total 35 min, the membranes were incubated with secondary anti-rabbit IgG-HRP conjugates at room temperature for another 1 h and then washed three times with TBST. The blots were visualized with Amersham ECL Plus western blotting detection reagents following the instructions of the manufacturer. Afterwards, the blots were stripped and reprobed with β-actin or GAPDH-specific antibody to ensure equal protein loading.
ROS detection
The cells were seeded in coverglass bottom dishes or 3.5 cm Petri dishes and cultured until 80–90% confluent and then treated with agents as indicated for 4 h. The treated cells were washed with phosphate-buffered saline (PBS), and then the fluorescent dyes, CM-H2DCF-DA or HE, were applied for 20 min at 37 °C at final concentrations of 5 or 10 μM, respectively. After that, the cells were washed twice with PBS and observed under a laser scanning confocal microscope (Leica TCS NT, Heidelberg, Germany) or analyzed by fluorescence-activated cell sorting (FACS) flow cytometry with excitation at 488 nm and emission at 530 nm for CM-H2DCF-DA or excitation at 543 nm and emission at 580 nm for HE.
Statistical analysis
Data were expressed as the mean ± standard deviation. A Student’s t test was performed to reveal differences between two treatment groups, and p < 0.05 (unless otherwise stated) was considered statistically significant.
Results
Vanadium compounds induced more pronounced growth inhibition in hepatoma HepG2 cells than that in hepatic L02 cells
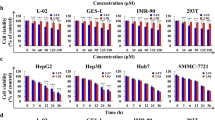
To determine whether vanadium compounds induce differential effects on the cell growth of human hepatoma HepG2 and hepatic L02 cells, MTT assays were performed after treatment with various concentrations of vanadate, VO(acac)2 or VO(ma)2 (50, 100 and 400 µM) for 72 h. As indicated in Fig. 1a, all three vanadium compounds led to a strong and dose-dependent suppression of viability in HepG2 cells, with an approximately 70% decrease in cell viability at a concentration of 100 μM. In contrast, a modest effect on L02 cells, with an ~20% decrease in cell viability, was observed with these vanadium compounds at the same concentration. To demonstrate the relationship between the decrease in cell viability and the inhibition of cell proliferation, the three-day growth curves of HepG2 and L02 cells in the presence or absence of vanadium compounds were analyzed, as shown in Fig. 1b, c, respectively. For HepG2 cells (Fig. 1b), 100 μM vanadate, VO(acac)2 or VO(ma)2 induced a significant reduction in cell viability after 48 h and resulted in stronger growth inhibition thereafter. For L02 cells (Fig. 1c), however, the inhibition by vanadium compounds was relatively insignificant.

Vanadium compounds exerted differential cell growth inhibition in HepG2 and L02 cells. a Cell viability comparison between HepG2 and L02 cells after treatment with vanadium compounds for 72 h. Cells were treated with increasing concentrations of vanadate, VO(acac)2 or VO(ma)2 for 72 h prior to the MTT assay. b, c Growth curves of HepG2 and L02 cells, respectively, treated with vanadium compounds. Cells were incubated with 100 μM vanadate, VO(acac)2 or VO(ma)2 on day 0, and the MTT assay was performed daily from day 0 to day 3. Error bars represent the standard deviation from triplicate runs (n = 3). The cell viability of each treatment group was compared to the corresponding untreated control, which was normalized to 100% cell viability. Statistical significances are indicated relative to the control HepG2 or L02 cells by *(p < 0.05) or **(p < 0.001)
These results showed that vanadium compounds induced more pronounced growth inhibition in HepG2 hepatoma cells.
Vanadium compounds blocked HepG2 cells in the G1/S phase but induced S and G2/M cell cycle arrest in L02 cells
To elucidate the mechanism of vanadium-induced growth inhibition described above, the cell cycle distribution was analyzed by flow cytometry. Although 100 μM vanadium compounds did not cause an obvious decrease in cell viability after 24 h in two cell lines (Fig. 1b, c), they did result in cell cycle arrest. Our previous studies have shown that they block the cell cycle of HepG2 at the G1/S stage [22]. By contrast, in L02 cells, as shown in Fig. 2a, all three compounds led to increased fractions in both the S and the G2/M phases. Kinetic analysis of cell cycle progression revealed a progressive accumulation of cells in the S and/or the G2/M phases in accordance with a continuous decrease in G0/G1 fraction (Fig. 2b). Figure 2c shows the concentration-dependent S and G2/M arrest in L02 cells.

Effects of vanadium compounds on cell cycle distribution and cell cycle regulatory proteins in HepG2 and L02 cells. a Representative flow cytometric profiles of the cell cycle distribution of L02 cells in the presence of vanadate (100 μM), VO(acac)2 (100 μM) and VO(ma)2 (200 μM) for 24 h. b Time courses of cell cycle arrest after treatment of L02 cells with various vanadium compounds at different time points. The concentrations of vanadium compounds were the same as indicated in a. c Dose-dependent effects on cell cycle distribution in L02 cells after treatment with various vanadium compounds for 24 h. d Dose-dependent effects of vanadate or VO(acac)2 on levels of cyclin D and phosphorylated pRb at Ser 780 or Ser 807/811 in HepG2 cells after 24 h of treatment. e Levels of phosphorylated Cdc2 at Tyr 15 in L02 cells treated with vanadate or VO(acac)2 for 24 h. The data shown for both flow cytometric analysis and western blots represent at least three independent experiments
The differential vanadium compound-induced cell cycle arrest in HepG2 and L02 cells was further evidenced by examining the key proteins implicated in regulating G1/S and G2/M transitions, respectively. As shown in Fig. 2d, upon treating HepG2 cells with increasing concentrations (50, 100, 200, 400 µM) of either V(V) (vanadate) or V(IV) (VO(acac)2) for 24 h, the levels of phosphorylated pRb at both Ser 780 and Ser 807/811 sites (hallmarks of the G1/S transition [25, 26]) were observed to decrease in a concentration-dependent manner. Moreover, the protein expression of cyclin D decreased concomitantly. In L02 cells, as shown in Fig. 2e, the level of phosphorylation of Cdc2 at Tyr 15 was significantly increased after treatment with 100 µM vanadate and VO(acac)2 for 24 h. The Cdc2–cyclinB complex, known as mitosis-promoting factor, is important for entry into mitosis [27]. Switching Cdc2 from an inactive to an active state requires dephosphorylation at the site of Tyr 15 [28]. Therefore, the increase in Cdc2 phosphorylation at the inactive Tyr 15 site clearly demonstrates that vanadium compounds can induce G2/M cell cycle arrest in L02 cells.
Signaling pathways involved in differential cell cycle regulation by vanadium compounds in HepG2 and L02 cells
Then we investigated how vanadium compounds caused different cell cycle arrests in the two cell lines. Although vanadium compounds were able to induce more pronounced growth inhibition in HepG2 cells, they still caused cell cycle arrest in L02 cells. To avoid possible damage to normal hepatic cells, the underlying mechanism must be investigated.
First, mitogen-activated protein kinase (MAPK) pathways were assessed, which are generally considered to play an important role in cell cycle regulation [6, 11, 29–32]. The activation states of the ERK, JNK and p38 signaling pathways with or without various concentrations of vanadate in HepG2 and L02 cells were measured after 24 h. As shown in Fig. 3a, vanadate dose-dependently activated the three pathways in HepG2 cells. In contrast, no apparent phosphorylation of these MAPKs was observed in L02 cells until a sufficient concentration of vanadium compounds was reached (400 μM). In other words, L02 cells exhibited a lower intensity of MAPK activation in response to vanadium compounds, which may explain their relative insensitivity to vanadium-induced growth inhibition. Similar results were obtained for VO(acac)2 (data not shown).

Vanadium compounds led to the differential activation of MAPK signal pathways in HepG2 and L02 cells. a Western blot analysis of phospho-ERK, phospho-JNK and phospho-p38 levels in HepG2 and L02 cells upon treatment with various concentrations of vanadate for 24 h. b Time courses of the effect of 100 μM vanadate on the phosphorylation of ERK, JNK, p38 and Cdc2 in L02 cells. c Involvement of ERK and JNK activation in vanadate-induced G2/M cell cycle arrest in L02 cells. Cells were exposed to 1 μM MEK inhibitor I and 20 μM SP600125 in the presence or absence of 100 μM vanadate for 24 h. Cells lysates were subjected to western blot analysis for phospho-Cdc2 (Tyr 15) and Cdc2. One out of at least three experiments with consistent results is shown
To further clarify whether vanadium compound-induced activation of MAPKs in L02 cells is time dependent, we examined the temporal effects of 100 μM vanadate on the phosphorylation of ERK, JNK and p38 kinases. After FBS starvation of L02 cells for 24 h, 10% FBS was restored in the presence or absence of 100 µM vanadate for 4, 8, 12, 16 and 24 h. As seen in Fig. 3b, vanadate highly increased the levels of phosphorylated ERK and JNK compared with the levels seen with exposure to 10% FBS at each time point. However, no phosphorylated p38 was observed. Moreover, after treatment with vanadate, the level of phosphorylated Cdc2 at its inactive site Tyr 15 was found to be significantly increased. This was in agreement with the time-dependent increase in the percentage of cells in the G2/M phase, as shown in Fig. 2b.
To determine the contributions of the ERK and JNK pathways induced by vanadium compounds to G2/M arrest in L02 cells, the influence of two inhibitors were evaluated: MEK inhibitor I for MEK-1 (upstream kinase for ERK), and SP600125 for JNK. As shown in Fig. 3c, co-incubation of vanadate with either MEK inhibitor I or SP600125 led to a remarkably decreased level of phosphorylated Cdc2, demonstrating that suppression of either the ERK or the JNK signaling pathway was able to abrogate Cdc2 inactivation by vanadate. This indicated that both the ERK and the JNK pathways were responsible for vanadium compound-induced G2/M cell cycle arrest in L02 cells.
This result was basically the same as that obtained for HepG2 cells. We previously reported that vanadium compounds can cause G1/S cell cycle arrest via the highly activated ERK pathway [22]. However, in that case, no obvious activation of JNK and p38 by low concentrations of vanadium compounds was observed at early time points. As the incubation time extended and the exposure concentration increased (Fig. 3a), all three pathways were activated. Therefore, it will be difficult to take advantage of the differential regulation of MAPK pathways in two cell lines by vanadium compounds in order to avoid possible damage to normal hepatic cells without decreasing their anticancer activities.
Vanadium compounds induced significant increases in ROS levels in L02 cells
Considering that many types of cancer cell have increased levels of reactive oxygen species (ROS) compared with their normal counterparts [33, 34], we speculated that differences between the redox states of the two cell lines could help to solve the problem. Therefore, we first evaluated the basal ROS levels in two cell lines. As shown in Fig. 4a, the mean DCF fluorescence, which indicates the level of H2O2 (a key component of oxidative stress), was about 20-fold lower in L02 cells than in HepG2 cells. We then examined whether treatment with vanadium compounds could induce ROS generation in L02 cells by detecting the intracellular levels of H2O2 and O ·−2 using the fluorescent dyes CM-H2DCF-DA and HE, respectively. In contrast to the insignificant ROS elevation in HepG2 cells [22], both H2O2 and O ·−2 levels in L02 cells were significantly increased 4 h after exposure to either vanadate (V) or vanadyl (IV) compounds (Fig. 4b–d). Figure 4e shows that vanadate can induce ROS generation dose-dependently.

Vanadium compounds caused significant elevations of ROS levels in L02 cells. a Basal ROS levels in HepG2 and L02cells. After CM-H2DCF-DA staining for 20 min, flow cytometry was performed to elucidate the intracellular basal ROS levels of L02 and HepG2 cells. b L02 cells were exposed to 500 μM vanadate, 100 μM VO(acac)2 or 200 μM VO(ma)2 for 4 h. As a positive control, L02 cells were treated for 30 min with 500 μM H2O2. The level of H2O2 in each treatment group was then determined by flow cytometric analysis. c, d Typical confocal microscope images of H2O2 and O ·−2 generated inside the L02 cells treated (or not) with vanadium compounds. H2O2 and O ·−2 were stained by CM-H2DCF-DA (green) and HE (red), respectively. 1 and 5 show phase contrast images of the same visual field as in 2 and 6; 2 and 6 show the untreated control cells; 3 and 7 show cells treated with 500 μM vanadate for 4 h; 4 and 8 show cells treated with 100 μM VO(acac)2 for 4 h. e Cell cycle analysis of the dose-dependent effect of vanadate on the levels of H2O2 in L02 cells. Similar results were obtained in three independent experiments
Vanadium compound-induced G2/M cell cycle arrest in L02 cells was due to ROS generation
To further elucidate the contribution of ROS production in L02 cells to the G2/M cell cycle arrest caused by vanadium compounds, a general ROS scavenger, N-acetylcysteine (NAC), was used. As shown in Fig. 5a, treatment with NAC can significantly decrease ROS generation. Moreover, in the presence of 20 mM NAC, vanadate-induced growth inhibition was somewhat ameliorated (Fig. 5b). This protective effect of NAC was further evidenced by monitoring the level of cell cycle regulatory protein. As shown in Fig. 5c, NAC significantly decreased the inhibitory phosphorylation of Cdc2 at Tyr 15 that occurred after 24 h of treatment. Likewise, the dose-dependent effects of vanadate on the activation of ERK and JNK were also counteracted by co-treatment with NAC. Along with the data shown in Fig. 3d, it can be concluded that vanadate induces G2/M cell cycle arrest in L02 cells, at least in part, through the ROS-dependent activation of ERK and JNK pathways.

Vanadate induces G2/M cell cycle arrest in L02 cells through an ROS-dependent mechanism. a NAC inhibited vanadate-induced ROS generation in L02 cells. Cells were pretreated with 20 mM NAC for 30 min followed by 4 h of co-incubation with 400 μM vanadate. The level of H2O2 in each treatment group was measured by flow cytometric analysis. One of two independent experiments with comparable results is presented. b NAC treatment restored the decrease in cell viability of L02 cells in response to vanadate. L02 cells were treated with 400 μM vanadate for 24 h in the presence or absence of NAC. The data were expressed as the mean ± standard deviation of triplicate counts (n = 3). Asterisks refer to statistically significant differences compared with the untreated control (normalized to 1), p < 0.05. Hash refers to a significant difference between the indicated groups (p < 0.01). c The levels of phosphorylated Cdc2, ERK and JNK after treatment with increasing concentrations of vanadate for 24 h in the absence or presence of 20 mM NAC. Representative blots of at least three independent experiments are shown
The antioxidant NAC did not attenuate the antitumor activity of vanadate in HepG2 hepatoma cells
The next issue to be investigated was whether the antioxidant NAC has any effect on the anticancer activities of vanadium compounds in HepG2 cells. As seen in Fig. 6a, incubation for 24 h with either 20 mM NAC or 400 μM vanadate alone resulted in significant growth suppression. Correspondingly, as shown in Fig. 6b, treatment with NAC caused a decrease in the basal ROS level in HepG2 cells. It was also noted that NAC itself led to a dose-dependently decreased level of phosphorylated pRb (Fig. 6c). Furthermore, although co-incubation with NAC and vanadate failed to exacerbate the decrease in cell viability induced by vanadate (Fig. 6a), a more pronounced reduction in pRb phosphorylation was observed when HepG2 cells were co-treated with vanadate and NAC for 24 h (Fig. 6c). Since the dephosphorylation converts pRb to its active state and exerts growth suppressive effects [35], the above results may at least indicate that NAC does not attenuate the antitumor activity of vanadate in HepG2 hepatoma cells.

Effect of NAC on the antitumor activity of vanadate towards HepG2 cells. a Effect of NAC on the viability of vanadate-treated HepG2 cells. The same method was used as described in Fig. 5b. Data are depicted as -fold increases compared to the untreated control (normalized to 1), for triplicate counts (n = 3). *p < 0.05. b NAC decreased the basal ROS level in HepG2 cells. The DCF fluorescence intensities of HepG2 cells treated with or without 20 mM NAC for 4 h were measured by flow cytometry. One of at least three experiments with consistent results is shown. c Dose-dependent effects of NAC on the phosphorylation levels of pRb at Ser 780 and Ser 807/811 in HepG2 cells treated with or without vanadate. Representative blots of at least three independent experiments are shown
Discussion
The present study showed the selectivity of vanadium compounds for human hepatoma HepG2 cells. Combined with our previous work [22], the differential mechanisms underlying vanadium compound-induced cell cycle arrest in HepG2 and L02 cells are proposed (Fig. 7).

Possible mechanisms underlying vanadium compound-induced cell cycle arrest in HepG2 and L02 cells
As illustrated in Fig. 7, it can be seen that, in contrast to HepG2 cells, all three of the vanadium compounds in either the V(V) or the V(IV) oxidation state, and as either simple salts or vanadium complexes with organic ligands, can induce significant ROS generation in L02 cells. Unexpectedly, vanadium compounds were not observed to cause significant ROS elevation in HepG2 cells. This may be attributed to the constitutive ROS production in the cells [36], which agrees with our results as seen in Fig. 4a. Under persistent conditions of intrinsic oxidative stress, HepG2 cells may become well adapted to such stress and develop an enhanced endogenous antioxidant capacity that makes them resistant to exogenous oxidative stress. Thus, even if vanadium compounds cause an increase in ROS generation, this increase may still lie within the cellular tolerability threshold and result in cellular adaptation, which may present as proliferation for example. It indicates that, in contrast to L02 cells, the growth of HepG2 cells may be more dependent on the level of ROS. This hypothesis was supported by the current study. As shown in Fig. 6a, b, NAC caused a decrease in cell viability in HepG2 cells, corresponding to a decrease in ROS level upon treatment with NAC. However, under the same conditions, NAC itself did not cause damage to L02 cells (Fig. 5b); indeed, it was shown to protect L02 cells from cytotoxicity caused by vanadium compounds.
It should be pointed out that, NAC, as a precursor of l-cysteine and reduced glutathione, is a thiol-containing antioxidant and is considered to be able to reduce the accumulation of ROS and oxidative damage to alleviate the toxicity of vanadium compounds to L02 cells. However, it was also suggested that NAC may react directly with vanadium compounds [37, 38]. For example, it is possible that NAC can act as a reducing agent, leading directly to the reduction of vanadate, or it may act as a ligand and form a complex with vanadate [38]. Recently, it was demonstrated that, at neutral pH, vanadate can form complexes with thiols without undergoing reduction [39]. Therefore, in the present study, to check whether an ROS-mediated mechanism occurs in vanadate-induced cytotoxicity in L02 cells, the possibility of a direct interaction between NAC and vanadate should be taken into account. Nevertheless, we can exclude this based on the effect of NAC on HepG2 cells. In the presence of vanadate, co-treatment with 20 mM NAC did not decrease the cytotoxicity of vanadate (Fig. 6a), demonstrating that the addition of NAC does not affect the effective concentration of vanadate by reacting with it. Anyway, no matter which mechanism operates, the application of vanadate with NAC as a synergistic agent did protect L02 cells but caused no significant effect on HepG2 cells.
Regarding the signaling pathway that mediates the differential effects of vanadium compounds on the two cell lines, it is worth noting that, in both cell lines, sustained overactivation of ERK contributed to cell cycle arrest by vanadium compounds, which is consistent with the increasing evidence indicating a link between overactivation of ERK and cell death [22, 31, 40, 41]. In addition, the current study further demonstrated that the two cell lines may use different mechanisms—one is ROS dependent and the other is ROS independent—to mediate ERK activation. There is thus the possibility that we could use antioxidants to modulate the differential responses of hepatoma cancer and normal liver cells to vanadium compounds. However, for a given type of cancer cell, the role of ROS in the regulation of biological processes is very complicated. Some researchers have proposed developing a combined set of parameters to indicate the potential of a cell type for ROS-manipulating therapies [42]. For vanadium compounds, although there was no significant increase in ROS generation in HepG2 cells, there is mounting evidence that they can induce ROS generation in many types of cancer cells [10–12, 43, 44]. Therefore, the effects of vanadium compounds may be cell specific, which means that a result obtained from one type of cell line cannot be simply extended to another situation. However, if the specific conditions used in an experiment are known and categorized, including the redox status and even the expressions of redox-sensitive transcription factors, personalized treatment may become possible, and thus we could optimize the performance of vanadium compounds. As we know, hepatocellular carcinoma has become the third most common cause of cancer-related death worldwide, and its incidence is increasing [45, 46]. Systemic chemotherapy has not been efficacious for it up to now [47]. Our results show that vanadium compounds can induce cell cycle arrest in HepG2 cells via an ROS-independent pathway, and that by using antioxidants as synergistic agents, damage to normal liver cells may be avoided, indicating the possibility of developing vanadium compounds as a therapeutic agent against this devastating disease. Further studies are needed to conclusively demonstrate the potential application of vanadium compounds.
Additionally, diabetes has been shown to be a risk factor for hepatocellular carcinoma as well as endometrial, colorectal and breast cancer [48, 49]. Vanadium compounds are well-known insulin-mimetic or -enhancing agents [50–52] and are also used as antitumor agents [1, 22, 53, 54]. The link between diabetes and cancer indicates that use of vanadium compounds in the treatment of diabetic patients with cancer may be beneficial. On the one hand, when vanadium compounds are applied in diabetes therapy, they exert cytostatic effects on cancer cells. On the other hand, as oxidative stress plays a crucial role in the development of insulin resistance and diabetes-related complications [55, 56], drugs applied in diabetes therapy are expected to normalize blood glucose concentration as well as to exhibit antioxidative properties. There are concerns over the use of vanadium compounds in diabetes treatment relating to their tendency to induce oxidative stress in various biological systems [1, 10–12, 57]. Therefore, it seems that the combined administration of vanadium and an antioxidant might also be beneficial for diabetes therapy.
In summary, the results presented in this study demonstrate that vanadium compounds significantly inhibit the proliferation of human hepatoma HepG2 cells but only have a modest effect on immortalized hepatic L02 cells, indicating the selectivity of vanadium compounds for tumor cells. Moreover, it was found that there are no essential distinctions between vanadium compounds with different oxidation states in terms of their actions on the two cell lines. Treatment with the antioxidant NAC was found to ameliorate the cytotoxicity of vanadium compounds in L02 cells, but it did not decrease their cytotoxicity in HepG2 cells. These results offer a better understanding of how vanadium compounds modulate cancer cells and their normal counterparts, and will be helpful in the therapeutic application of vanadium compounds along with antioxidants as synergistic agents.
Abbreviations
- Akt/PKB:
-
Protein kinase B
- Cdc2/Cdk1:
-
Cell division control protein 2 homolog/cyclin dependent kinase 1
- CM-H2DCF-DA:
-
5-(and-6)-Chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
- ERK:
-
Extracellular signal-regulated protein kinase
- FBS:
-
Fetal bovine serum
- HE:
-
Dihydroethidium
- JNK:
-
c-Jun N-terminal kinase
- MAPK:
-
Mitogen-activated protein kinase
- MTT:
-
3-(4,5-Dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide
- NAC:
-
N-acetylcysteine
- NaVO3 :
-
Sodium metavanadate
- PI3-K:
-
Phosphatidylinositol 3-kinase
- pRb:
-
Retinoblastoma tumor suppressor protein
- ROS:
-
Reactive oxygen species
- VO(acac)2 :
-
Bis(acetylacetonato)-oxovanadium(IV)
- VO(ma)2 :
-
Bis(maltolato)-oxovanadium(IV)
References
Evangelou AM (2002) Crit Rev Oncol Hematol 42:249–265
Ress NB, Chou BJ, Renne RA, Dill JA, Miller RA, Roycroft JH, Hailey JR, Haseman JK, Bucher JR (2003) Toxicol Sci 74:287–296
Hwang JT, Lee M, Jung SN, Lee HJ, Kang I, Kim SS, Ha J (2004) Carcinogenesis 25:2497–2507
Tang H, Sun Y, Xiu Q, Lu H, Han H (2007) Arch Biochem Biophys 468:92–99
Zhang Z, Gao N, He H, Huang C, Luo J, Shi X (2004) Mol Cell Biochem 255:227–237
Chien PS, Mak OT, Huang HJ (2006) Biochem Biophys Res Commun 339:562–568
Goc A (2006) Cent Eur J Biol 1:314–332
Desoize B (2004) Anticancer Res 24:1529–1544
Molinuevo MS, Barrio DA, Cortizo AM, Etcheverry SB (2004) Cancer Chemother Pharm 53:163–172
Gao N, Ding M, Zheng JZ, Zhang Z, Leonard SS, Liu KJ, Shi X, Jiang BH (2002) J Biol Chem 277:31963–31971
Zhang Z, Leonard SS, Huang C, Vallyathan V, Castranova V, Shi X (2003) Free Radic Biol Med 34:1333–1342
Zhang Z, Huang C, Li J, Leonard SS, Lanciotti R, Butterworth L, Shi X (2001) Arch Biochem Biophys 392:311–320
Li M, Ding W, Baruah B, Crans DC, Wang R (2008) J Inorg Biochem 102:1846–1853
Figiel I, Kaczmarek L (1997) Neuroreport 8:2465–2470
Chin LS, Murray SF, Harter DH, Doherty PF, Singh SK (1999) J Biomed Sci 6:213–218
Gamero AM, Larner AC (2001) J Biol Chem 276:13547–13553
Tracey AS, Willsky GR, Takeuchi E (2007) Vanadium: chemistry, biochemistry, pharmacology and practical applications. CRC, Boca Raton
Trachootham D, Alexandre J, Huang P (2009) Nat Rev Drug Discov 8:579–591
Thompson KH, Orvig C (2000) J Chem Soc Dalton Trans 2885–2892
Yuen VG, Caravan P, Gelmini L, Glover N, McNeill JH, Setyawati IA, Zhou Y, Orvig C (1997) J Inorg Biochem 68:109–116
Crans DC (2000) J Inorg Biochem 80:123–131
Fu Y, Wang Q, Yang XG, Yang XD, Wang K (2008) J Biol Inorg Chem 13:1001–1009
Natarajan M, Mohan S, Martinez BR, Meltz ML, Herman TS (2000) Cancer Detect Prev 24:405–414
Funk D, Schrenk H-H, Frei E (2007) Biotechniques 43:178–182
Sherr CJ (1996) Science 274:1672–1677
Reed SI (1997) Cancer Surv 29:7–23
Morgan DO (1995) Nature 374:131–134
Dunphy WG (1994) Trends Cell Biol 4:202–207
Garcia Z, Kumar A, Marques M, Cortes I, Carrera AC (2006) EMBO J 25:655–661
Meloche S, Pouyssegur J (2007) Oncogene 26:3227–3239
Chambard JC, Lefloch R, Pouyssegur J, Lenormand P (2007) Biochim Biophys Acta 1773:1299–1310
Jaspers I, Samet JM, Erzurum S, Reed W (2000) Am J Respir Cell Mol Biol 23:95–102
Toyokuni S, Okamoto K, Yodoi J, Hiai H (1995) FEBS Lett 358:1–3
Szatrowski TP, Nathan CF (1991) Cancer Res 51:794–798
Tamrakar S, Rubin E, Ludlow JW (2000) Front Biosci 5:D121–D137
Lou YW, Chen YY, Hsu SF, Chen RK, Lee CL, Khoo KH, Tonks NK, Meng TC (2008) FEBS J 275:69–88
Baran Enrique J (2003) J Braz Chem Soc 14:878–888
Legrum W (1986) Toxicology 42:281–289
Crans DC, Zhang B, Gaidamauskas E, Keramidas AD, Willsky GR, Roberts CR (2010) Inorg Chem. doi:10.1021/ic100080k
Wang YF, Jiang CC, Kiejda KA, Gillespie S, Zhang XD, Hersey P (2007) Clin Cancer Res 13:4934–4942
Javvadi P, Segan AT, Tuttle SW, Koumenis C (2008) Mol Pharmacol 73:1491–1501
Wang J, Yi J (2008) Cancer Biol Ther 7:1875–1884
Yang XG, Yang XD, Yuan L, Wang K, Crans DC (2004) Pharm Res 21:1026–1033
Capella MA, Capella LS, Valente RC, Gefe M, Lopes AG (2007) Cell Biol Toxicol 23:413–420
Montalto G, Cervello M, Giannitrapani L, Dantona F, Terranova A, Castagnetta LA (2002) Ann N Y Acad Sci 963:13–20
Sangiovanni A, Del Ninno E, Fasani P, De Fazio C, Ronchi G, Romeo R, Morabito A, De Franchis R, Colombo M (2004) Gastroenterology 126:1005–1014
Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, Tovar V, Roayaie S, Minguez B, Sole M, Battiston C, Van Laarhoven S, Fiel MI, Di Feo A, Hoshida Y, Yea S, Toffanin S, Ramos A, Martignetti JA, Mazzaferro V, Bruix J, Waxman S, Schwartz M, Meyerson M, Friedman SL, Llovet JM (2008) Gastroenterology 135:1972–1983
Davila JA, Morgan RO, Shaib Y, McGlynn KA, El-Serag HB (2005) Gut 54:533–539
Dann SG, Selvaraj A, Thomas G (2007) Trends Mol Med 13:252–259
Thompson KH, Orvig C (2006) J Inorg Biochem 100:1925–1935
Thompson KH, Lichter J, LeBel C, Scaife MC, McNeill JH, Orvig C (2009) J Inorg Biochem 103:554–558
Sakurai H, Yoshikawa Y, Yasui H (2008) Chem Soc Rev 37:2383–2392
Faneca H, Figueiredo VA, Tomaz I, Goncalves G, Avecilla F, Pedroso de Lima MC, Geraldes CF, Pessoa JC, Castro MM (2009) J Inorg Biochem 103(4):601–608
Etcheverry SB, Ferrer EG, Naso L, Rivadeneira J, Salinas V, Williams PA (2008) J Biol Inorg Chem 13:435–447
Evans JL, Goldfine ID, Maddux BA, Grodsky GM (2002) Endocr Rev 23:599–622
Houstis N, Rosen ED, Lander ES (2006) Nature 440:944–948
Oster MH, Llobet JM, Domingo JL, German JB, Keen CL (1993) Toxicology 83:115–130
Acknowledgments
This work was supported by the National Natural Science Foundation of China (20871008) and the Scientific Research Foundation for Returned Overseas Chinese Scholars, State Education Ministry.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Q., Liu, TT., Fu, Y. et al. Vanadium compounds discriminate hepatoma and normal hepatic cells by differential regulation of reactive oxygen species. J Biol Inorg Chem 15, 1087–1097 (2010). https://doi.org/10.1007/s00775-010-0668-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-010-0668-4