
Abstract
Nitrile hydratases (NHase), which catalyze the hydration of nitriles to amides, have an unusual Fe3+ or Co3+ center with two modified Cys ligands: cysteine sulfininate (Cys-SO2 −) and either cysteine sulfenic acid or cysteine sulfenate [Cys-SO(H)]. Two catalytic mechanisms have been proposed. One is that the sulfenyl oxygen activates a water molecule, enabling nucleophilic attack on the nitrile carbon. The other is that the Ser ligand ionizes the strictly conserved Tyr, activating a water molecule. Here, we characterized mutants of Fe-type NHase from Rhodococcus erythropolis N771, replacing the Ser and Tyr residues, αS113A and βY72F. The αS113A mutation partially affected catalytic activity and did not change the pH profiles of the kinetic parameters. UV–vis absorption spectra indicated that the electronic state of the Fe center was altered by the αS113A mutation, but the changes could be prevented by a competitive inhibitor, n-butyric acid. The overall structure of the αS113A mutant was similar to that of the wild type, but significant changes were observed around the catalytic cavity. Like the UV–vis spectra, the changes were compensated by the substrate or product. The Ser ligand is important for the structure around the catalytic cavity, but is not essential for catalysis. The βY72F mutant exhibited no activity. The structure of the βY72F mutant was highly conserved but was found to be the inactivated state, with αCys114-SO(H) oxidized to Cys-SO2 −, suggesting that βTyr72 affected the electronic state of the Fe center. The catalytic mechanism is discussed on the basis of the results obtained.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nitrile hydratase (NHase; EC 4.2.1.84) is the key enzyme in the bacterial nitrile degradation system and catalyzes the hydration of nitriles to the corresponding amides [1–3]. NHase is used for industrial production of acrylamide (more than 30,000 tons per year) and nicotinamide, and it is considered the most successful biocatalyst. NHase consists of α and β subunits and contains a low-spin non-heme Fe3+ [4] or non-corrin Co3+ [5] catalytic center. Because of high amino acid sequence similarities, the catalytic mechanism is believed to be conserved. Fe-type NHase is relatively unstable, and a competitive inhibitor, n-butyric acid (n-BA), is commonly used as a stabilizing agent for its purification, storage, and various spectroscopic measurements [6]. Fe-type NHases from Rhodococcus erythropolis N771 (ReNHase) and Comamonas testosteroni Ni1 (CtNHase) are transformed into a stable inactive state by nitrosylation of the Fe center in the dark and are immediately reactivated by photoinduced denitrosylation [7–9].
The crystal structures in this family display a high degree of similarity, including the Fe-type [10, 11] and Co-type [12, 13] NHase proteins as well as a related protein, thiocyanate hydrolase (SCNase) [14]. The metal is bound in a distorted octahedral geometry within a strictly conserved motif of the α subunit, Cys1-Xxx-Leu-Cys2-Ser-Cys3. In Fe-type ReNHase, the ligand atoms are three sulfurs of αCys109, αCys112, and αCys114 (corresponding to Cys1, Cys2, and Cys3 in the motif), the two main chain amide nitrogens of αSer113 and αCys114, and an exogenous NO molecule in the nitrosylated state [11]. The sixth ligand was found to be a water molecule in the Co-type NHase from Pesudonocardia thermophila JCM 3095 (PtNHase) [12] and an anion in SCNase [14]. αCys112 and αCys114 of ReNHase are posttranslationally modified to cysteine sulfinic acid (Cys-SO2H) and cysteine sulfenic acid (Cys-SOH), respectively [11], and both Cys modifications are conserved in Co-type NHase [12] and in SCNase [14]. Fourier IR spectroscopy [15] and K-edge X-ray absorption spectroscopy [16] of ReNHase in combination with density functional theory calculations revealed that αCys112-SO2H was deprotonated as Cys-SO2 −, whereas the protonation state of αCys114-SOH remained uncertain [Cys-SO(H)] because it was assigned as Cys-SO− by Fourier transform IR spectroscopy but was demonstrated to be protonated by X-ray absorption spectroscopy. The sulfinyl and sulfenyl groups protrude into the catalytic cavity together with the hydroxyl group of the serine ligand to form a “claw-setting” structure [11]. The most widely believed catalytic mechanism involves direct coordination of nitriles to the metal. In this mechanism, the nitrile carbon increases its electrophilicity by coordinating to the Lewis acidic metal and then is attacked by a water molecule, which is activated by a “general base” via an outer-sphere mechanism [17–19]. From the crystal structures of Fe-type [10, 11] and Co-type [12, 13] NHases, two oxidized Cys ligands, the Ser ligand, and a strictly conserved Tyr residue [corresponding to αCys112-SO2 −, αCys114–SO(H), αSer113, and βTyr72 in ReNHase, respectively] can be considered as candidates for the “general base.”
ReNHase reconstituted anaerobically from unmodified subunits was found to display its catalytic activity only after oxygenation of the Cys ligands [20]. Specific oxidation of the Cys-SO(H) ligand to Cys-SO2 − resulted in irreversible inactivation [21]. In some model complexes mimicking NHase active sites, nitrile hydration activity was elevated by oxygenation of the thiolato ligands [22–24]. Extensive studies on NHase model complexes containing sulfurs in different oxidation states have suggested that sulfur oxidation fine-tuned the Lewis acidity of the metal center [16, 24–26] and/or that the sulfenyl oxygen exhibited a nucleophilic character [24, 26]. We recently performed time-resolved X-ray crystallographic analyses of ReNHase [27] using a novel substrate, tert-butyl isonitrile (t-BuNC), which was hydrolyzed to tert-butylamine and carbon monoxide [27, 28]. From monitoring of time-resolved crystal structures of the NHase–substrate complex, we proposed a catalytic mechanism in which the substrate coordinated to the metal was attacked by a water molecule activated by Oδ of αCys114-SO(H) (Fig. 1a). The same reaction model was proposed by a density functional theoretical calculation study based on the first-shell mechanism [29]. Very recently, we found that the recombinant SCNase having only the Cys2-SO2 − modification exhibited very weak catalytic activity but was activated by the monooxygenation of γCys131, confirming the importance of the sulfenyl group of γCys131-SO(H) in SCNase [30].

Proposed catalytic mechanisms of nitrile hydratase (NHase). a A water molecule activated by the sulfenate group of αCys114-SO(H) performs nucleophilic attack on the nitrile carbon. The model was based on [27]. b The serine ligand [corresponding to αSer113 of NHase from Rhodococcus erythropolis N771 (ReNHase)] functions as the general base and ionizes the phenolate oxygen of the strictly conserved adjacent tyrosine residue (corresponding to βTyr72 of ReNHase), which activates a water molecule. The model was based on [33]
In contrast, Mitra and Holz [31] studied the pH and temperature dependence of the kinetic parameters of PtNHase in detail, and they concluded that the Ser ligand (αSer112 in PtNHase) and two adjacent residues (βTyr68 and βTrp72) formed a catalytic triad to function as a catalytic base. The βTyr68 residue corresponds to βTyr72 in ReNHase, and its substitution with Phe in PtNHase resulted in an elevated K m value and a significantly decreased k cat value [32]. Moreover, the same analyses were performed on CtNHase. The amino acid replacement of Trp, which corresponded to βTrp72 of PtNHase, by Val led to the suggestion of a model where αSer112 and βTyr68 activated a water molecule that attacked the nitrile carbon (Fig. 1b) [33].
The reaction mechanism of NHase remains controversial. To study the roles of the Ser ligand and the Tyr residue and thus to test the model, we constructed mutant ReNHases with mutations of αSer113 and βTyr72 to Ala and Phe, respectively, and examined the kinetic parameters, UV–vis absorption spectra, and crystal structures. The results clearly indicated that the hydroxyl group of αSer113 affected the electronic state of the Fe center as well as the structure around the catalytic cavity including the hydrogen-bond networks, but was not essential for catalytic activity. On the basis of the results, the catalytic mechanism is discussed.
Materials and methods
Generation of the mutant NHases
The expression plasmids pRCN103, containing the genes for the NHase α and β subunits and the NHase activator, and pHSGβ, containing the NHase β subunit gene [34], were used for mutagenesis. Site-directed mutagenesis was performed using a QuikChange site-directed mutagenesis kit (Stratagene, CA, USA). pRCNαS113A, pRCNβY72F, and pHSGβY72F were constructed. All mutations were verified by DNA sequencing using an ABI 3130 genetic analyzer (Applied Biosystems, CA, USA). pRCNαS113A + pHSGβ and pRCNβY72F + pHSGβY72F were used for the expression of the αS113A and βY72F mutant NHases, respectively. The expression of each mutant NHase was performed in Escherichia coli HMS174(DE3)pLysS harboring the set of plasmids, as previously described [35]. The harvested E. coli cells were resuspended in 150 mL of 50 mM tris(hydroxymethyl)aminomethane (Tris)–HCl, pH 7.5, and a spontaneous NO donor, 1-hydroxy-2-oxo-3-(N-methyl-3-aminopropyl)-3-methyl-1-triazene (Dojindo, Kumamoto, Japan), was added at a final concentration of 0.25 mM under an argon atmosphere. The cells were incubated at 27 °C in a water bath for 1 h in the dark and were harvested by centrifugation (10,800g for 10 min). The NO-treated wild-type and mutant NHases were purified in the dark, as described previously [36]. The purified enzymes were stored in 50 mM Tris–HCl, pH 7.5, at −80 °C in the dark at a concentration of 20 mg/mL. Photoinduced denitrosylation was carried out by illumination using a photoreflector lamp (500 W SPOT, Toshiba, Tokyo, Japan) on ice for 15 min. Protein concentrations were determined by the absorbance at 280 nm (ε 280 of the purified NHase in the active state was 1.5 mL/mg/cm and in the nitrosylated state was 1.7 mL/mg/cm).
Assay of NHase activity
NHase activity was determined using reversed-phase high-performance liquid chromatography (HPLC). Photodenitrosylated NHase was dissolved in 50 mM sodium phosphate, pH 7.5, containing 50 mM n-BA at a protein concentration of 19 μM. Then, 0.5 μL of the NHase solution was added to 500 μL of the assay mixture, 50 mM sodium phosphate, pH 7.5, containing methacrylonitrile at concentrations of 0, 0.25, 0.50, 1.0, 1.5, 2.0, or 2.5 mM, and the resulting mixture was incubated for 1 min at room temperature. To determine the pH profile of the kinetic parameters, 50 mM 2-morpholinoethanesulfonic acid–NaOH (pH 6.0–6.5) and 50 mM Tris–HCl (pH 7.0–8.5) were used, respectively. The reaction was terminated by the addition of 500 μL of 3 M HCl, and the protein was removed by centrifugation at 20,400g for 10 min. Next, 100 μL of the supernatant fraction was analyzed by its absorbance at 224 nm using reversed-phase HPLC at a flow rate of 1.0 mL/min on ODS-80TS (4.6 mm × 150 mm; Tosoh, Tokyo, Japan), connected to a Waters 2690 HPLC system (Waters, Milford, MA, USA). Solvent A was 5 mM H3PO4, pH 2.9, and solvent B was CH3CN (Sigma-Aldrich, MO, USA). The column was equilibrated with 99% solvent A and 1% solvent B, and the samples were eluted isocratically with the same buffer. The kinetic parameters were determined from the [S/v]–[S] plot [37]. One unit of activity was defined as the quantity of NHase that produced 1 μmol of methacrylamide per minute.
Crystallization of the mutant ReNHases
Crystals of the NO-treated mutant ReNHases were grown using the vapor-diffusion hanging-drop method at 20 °C in the dark. Two microliters of the NO-treated mutant ReNHase (20 mg/mL protein in 50 mM Tris–HCl, pH 7.5) was mixed with an equal volume of the precipitant solution (20% PEG8000, 0.10 M Tris–HCl, pH 7.5, 0.30 M MgCl2) and equilibrated against 0.40 mL precipitant solution. Crystals with dimensions of about 0.4 × 0.3 × 0.3 mm3 grew within 1 day in the dark at 20 °C. Photoinduced denitrosylation of NHase in crystals was performed using a cold light illumination system (10,000 lx; LG-PS2, OLYMPUS, Tokyo, Japan).
Preparation of the crystals of the wild-type and αS113A mutant NHases in complex with tert-butylnitrile
Crystals of the wild-type and nitrosylated αS113A mutant were first vapor-soaked with cryoprotectant solution (30% PEG8000, 0.10 M Tris–HCl, pH 7.5, 0.60 M MgCl2) for 1 day. They were then vapor-soaked for 1 day with a mother liquor solution containing tert-butylnitrile (t-BuCN) at a final concentration of 0.10 M. After being mounted, the enzymes in the crystals were activated by light-induced denitrosylation (10,000 lx) with a cold light illumination system (LG-PS2, OLYMPUS, Tokyo, Japan), and the reaction proceeded for 90 min in the wild type, and for 60 and 90 min in the αS113A mutant at 20 °C. At each elapsed time, the reaction was terminated by flash-cooling with N2 gas at −178 °C.
X-ray data collection, structure determination, and refinements
Diffraction data were collected using a Quantum 315 CCD detector (ADSC) at beamline BL-5A (λ = 1.000 Å) of the Photon Factory (Tsukuba, Japan) at −178 °C. Each data set was indexed, merged, and scaled using the HKL2000 program suite [38]. All mutant ReNHase crystals belonged to the C2 space group. One heterodimer of α and β subunits populated the asymmetric unit. Molecular replacement was performed with MOLREP [39] in the CCP4 program suite [40], using the structure of the nitrosylated ReNHase in the P21212 space group [Protein Data Bank (PDB) ID 2AHJ] [11] as the initial coordinates. The models obtained were improved by iterative cycles of crystallographic refinement using REFMAC5 [41] for αS113A active, αS113A + product, and wild type + product and using SHELX97 [42] for αS113A + substrate and βY72F, and by manual model rebuilding using Coot [43]. Models were cross-validated by the SigmaA-weighted electron density maps [44] calculated with both 2mFobs − DFcalc and mFobs − DFcalc coefficients. The refinements were performed using a maximum likelihood target with bulk solvent corrections. During the structure refinement, approximately 5% of the amplitude data were set aside to monitor the progress of refinement using the R free factor. Solvent water molecules were gradually introduced if the peaks that were contoured at more than 4.0σ in the mFobs − DFcalc electron density were in the range of a hydrogen bond. The tert-butyl groups of t-BuCN were manually fit into the resultant difference electron density map, and their coordinate data were then refined using REFMAC5 [41]. Data collection and refinement statistics are summarized in Table 1. All structural figures were generated using PyMOL (http://pymol.sourceforge.net/).
PDB accession codes
The atomic coordinates and structure factors of the wild-type protein in complex with trimethylacetamide (t-BuCONH2), the αS113A mutant in the active state, the αS113A mutant in the nitrosylated inactive state in complex with t-BuCN, the αS113A mutant in the active state in complex with t-BuCONH2, and the βY72F mutant ReNHase have been deposited in the PDB under accession codes 3A8O, 3A8L, 3A8G, 3A8H, and 3A8M, respectively.
Results and discussion
Kinetic analysis of mutant NHases
Wild-type, αS113A mutant, and βY72F mutant NHases were expressed in E. coli, treated with NO to convert to stable nitrosylated forms, and then purified in the dark [7, 8]. The NHase activities of the αS113A mutant before and after light illumination were 0.0 and 8.6 × 102 units/mg protein, respectively, when 2.5 mM methacrylonitrile was used as the substrate. In contrast, the βY72F mutant exhibited no catalytic activity regardless of light illumination. The kinetic parameters for the αS113A mutant at pH 7.5 are listed in Table 2. The K m and k cat values of the αS113A mutant were 1.3 mM and 6.5 × 102 s−1. This K m is 1.7 times larger than that of the wild type, and k cat decreased by approximately 40% relative to that of the wild type. These results indicate that the hydroxyl group of αSer113 is not essential for the catalytic mechanism of NHase. The apparent pH for the maximal k cat for the αS113A mutant was 7.5. It was the same as that for the wild type (Fig. S1).
UV–vis absorption spectra of the mutant NHases
Figure 2 shows the UV–vis absorption spectra of the wild-type, αS113A mutant, and βY72F mutant NHases before and after light illumination in the absence and presence of 40 mM n-BA. Wild-type NHase in the nitrosylated state showed the same spectra with a shoulder peak at 370 nm, which had been assigned as an NO → Fe charge transfer transition [8], both in the absence (Fig. 2a) and in the presence (data not shown) of n-BA. Upon photoinduced denitrosylation, the 370-nm peak decreased, and a S → Fe charge transfer band at 680 nm (without n-BA) or at 710 nm (with n-BA) appeared [36] (Fig. 2a). In the absence of n-BA, the αS113A mutant exhibited a broad absorption from 300 to 800 nm both before and after light illumination (Fig. 2b). The shoulder peak at 370 nm was observed before light illumination, but no S → Fe charge transfer peak was apparent near 680 nm after light illumination. Interestingly, in the presence of n-BA, the αS113A mutant showed spectral features similar to those of the wild-type both before and after light illumination. These results indicate that the electronic state of the Fe center is significantly altered by the removal of the hydroxyl group of αSer113 in the absence of n-BA, but the alterations could be prevented by the presence of n-BA. The spectrum of the βY72F mutant exhibited a very weak shoulder peak at 370 nm and a peak at 698 nm before light illumination both in the absence and in the presence of n-BA (Fig. 2c). The spectra were unchanged after light illumination (data not shown). The spectral features are very similar to those of the denitrosylated NHase, which is inactivated by specific further oxygenation of αCys114-SO(H) to Cys-SO2H [21].

UV–vis absorption spectra of a wild-type, b αS113A mutant and c βY72F mutant NHases. The black lines and red lines indicate the spectra before and after light illumination in the absence of n-butyric acid (n-BA), whereas the blue lines and green lines indicate the spectra before and after light illumination in the presence of 40 mM n-BA. The protein concentrations for the wild-type, αS113A mutant, and βY72F mutant NHases were 1.1, 0.74, and 1.0 mg/mL, respectively. The buffers used were 50 mM tris(hydroxymethyl)aminomethane–HCl, pH 7.5, in the absence or presence of 40 mM n-BA. All spectra were recorded using a Cary50 (Varian, CA, USA). The spectra were measured with a quartz cuvette (light path, 10 mm) at room temperature
Structure of the αS113A mutant in the active state
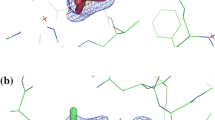
When crystals of the NO-treated αS113A mutant were used for structural determination, a water molecule rather than an NO molecule was observed in the sixth ligand site (data not shown). Since the structure of the wild-type protein was determined in the nitrosylated state under the same experimental conditions, the NO molecule is likely to be readily released by light. This explanation is consistent with our previous finding that the hydroxyl oxygen of αSer113 formed a claw setting with two oxygen atoms from the modified Cys ligands to stabilize the NO ligand [11]. To ensure light-induced denitrosylation, we illuminated crystals of the NO-treated αS113A mutant for 30 min before data collection. The overall structure was similar to that of the wild-type protein (PDB ID 2CYZ) except at the mutated site and around the Fe center. Figure 3 shows the electron density maps around the Fe center of the αS113A mutant after light illumination for 30 min. The average root-mean-square deviation (RMSD) between the αS113A mutant and the wild type was 0.13 Å for all atoms. The two Cys modifications, αCys112-SO2 − and αCys-SO(H), were conserved. The sixth ligand was a water molecule with an Fe–O(H2O) distance of 2.0 Å.

Structure around the Fe center of the αS113A mutant ReNHase in the photoinduced denitrosylated state. F o − F c omit electron density maps at 3.0σ contour (blue mesh) were superimposed on the refined structures. Green, blue, red, yellow, and orange sticks represent carbon, nitrogen, oxygen, and sulfur atoms, respectively
Figure 4a shows the superimposed structures around the Fe centers of the αS113A mutant and the wild type (PDB ID 2CYZ). In the αS113A mutant, the phenol groups of βTyr37 and βTyr72 are slightly displaced. The βTyr76 was found in two conformations with about equal ratio. The phenol group in conformer A was similar to that of the wild type, whereas in conformer B, it was displaced toward the substrate binding pocket by 6.0 Å (measured between the phenol oxygen atoms of conformers A and B). Two additional hydration water molecules, H2O(254) and H2O(551), were observed in the αS113A mutant (Fig. 4a). In the wild type (Fig. 5a), Oγ of αSer113 hydrogen-bonded with βTyr72 and H2O(102) and formed hydrogen-bond networks with βTyr37, βTyr72, αSer113, H2O(102), H2O(96), αTrp117, αThr115, and αGln90. In contrast, in the S113A mutant (Fig. 5b), H2O(254) was likely to be positioned at the space caused by the removal of Oγ of αSer113 and formed hydrogen bonds with βTyr37, βTyr72, H2O(551), and H2O(78) (corresponding to H2O(102) in the wild type) to stabilize the catalytic cavity, with the distinct conformation including βTyr37, βTyr72, and conformer B of βTyr76. Additionally, a disordered water molecule (H2O(2A) and H2O(2B) with the occupancy of 2A to 2B of 75:25) observed in the wild type disappeared in the αS113A mutant. H2O(2A) hydrogen-bonded with Oδ of αCys114-SO(H) as well as Nη2 of βArg56, which had previously been shown to be essential for catalytic activity [45], suggesting that the hydroxyl group of αSer113 affected the electronic state around αCys114-SO(H). The results suggested that the hydroxyl group of αSer113 affected not only the hydrogen-bond networks around the catalytic center but also the protein structure around the catalytic cavity.

Comparison of the structure around the catalytic cavity for the wild-type and αS113A mutant ReNHases. Stereo views of the superimposed structures around the Fe centers of a the αS113A mutant and the wild type (Protein Data Bank ID 2CYZ) in the active state, b the αS113A mutant in complex with t-BuCN and the wild type in complex with t-BuNC in the nitrosylated state (Protein Data Bank ID 2ZPE), and c the αS113A mutant in complex with t-BuCN and the wild type in complex with t-BuCN after 60 min (αS113A) and 90 min (wild type) of light illumination. Green, gray, blue, red, yellow, and orange sticks represent carbon (αS113A), carbon (wild type), nitrogen, oxygen, sulfur, and iron atoms, respectively. The coordinated water and hydration water molecules for αS113A and the wild type are shown as red and brown balls, respectively. A and B in the amino acid residues labels indicate α and β subunits, respectively. ACSD-112 and ACSO-114 represent αCys112-SO2 − and αCys114-SO(H), respectively

The hydrogen-bond networks around the catalytic cavity of a the wild-type and b the αS113A mutant NHases in the active state. Red, blue, yellow, and orange circles represent oxygen, nitrogen, sulfur, and iron atoms, respectively. The oxygen atoms that stand alone displayed hydration water molecules. The dotted lines and the dashed lines represent possible hydrogen bonds. Blue dotted lines represent the hydrogen bond observed in both the wild type and the αS113A mutant, whereas green dashed lines represent hydrogen bonds that disappeared or appeared in the mutant
Structures of the wild-type and αS113A mutant NHases in complex with substrate and product
To obtain the structure of wild-type NHase in complex with the product, crystals of nitrosylated wild-type NHase were soaked with a nitrile substrate, t-BuCN, at a final concentration of 0.10 M in the dark, and the crystalline enzymes were activated by light illumination for 90 min. The structure (Fig. 4c, gray stick) was found to be very similar to that of the nitrosylated NHase in complex with an isonitrile substrate, t-BuNC [27] (Fig. 4b, gray stick), except that the sixth NO ligand was replaced by a water molecule. A product molecule, t-BuCONH2, was observed in the catalytic cavity with no hydrogen-bonding contact to the protein moiety. When crystals of the wild-type NHase in the denitrosylated state were soaked with t-BuCONH2, it did not coordinate to the iron but positioned itself in the catalytic cavity, suggesting that the binding affinity of the product for the metal is not so strong (Y. Iijima et al., unpublished results). We note that, after 90 min of light illumination, many small crystals of t-BuCONH2 appeared around the NHase crystals, which showed that wild-type NHase catalyzed t-BuCN hydration repeatedly in its crystals.
We also soaked crystals of the NO-treated αS113A mutant with t-BuCN in the dark and determined the crystal structures before and after light illumination for 60 and 90 min. The structures of the αS113A mutant soaked with t-BuCN before and after light illumination for 60 min were superimposed with those of the wild type in complex with t-BuNC and t-BuCONH2 (Fig. 4b, c). Before light illumination, an NO molecule was observed at the sixth ligand site of the αS113A mutant in the same position as that of the wild type, confirming that the αS113A mutant is nitrosylated by NO treatment and denitrosylated by light illumination. The substrate t-BuCN is observed at the catalytic cavity (Fig. 4b). The position of the tert-butyl carbon was identical to that of t-BuNC in the structure of the nitrosylated wild type in complex with t-BuNC, although the directions of the molecules were different. After 60 min of illumination, the NO molecule was replaced by a water molecule, and electron densities corresponding to t-BuCN disappeared, whereas those of t-BuCONH2 were observed. Again, the position of the tert-butyl carbon of t-BuCONH2 in the αS113A mutant was identical to that in the wild type. The structure was unchanged even after 90 min of illumination (data not shown). The hydration water molecules as well as the conformation of the side chains around the Fe center were very similar between the αS113A mutant and the wild type when the substrate or the product was in the catalytic cavity (Fig. 4b, c). In the structures of the substrate and the product complexes, the RMSDs between the αS113A mutant and the wild type were 0.06 and 0.07 Å, which are dramatically smaller than the values using the substrate-free active-state structure. In accordance with the results of the UV–vis spectral measurements, the structural changes induced by the loss of the hydroxyl group of αSer113 were compensated by the substrate or product binding. The reason for the conservation of the structure around the catalytic cavity including hydration water molecules as well as the UV–vis absorption spectral features of the Fe center when the substrate or n-BA was added is not known. The occupation of active-site pocket by the substrate, product, or n-BA may drive βTyr76 to populate conformer A by steric hindrance, and thus the hydrogen-bond networks around the active-site pocket as well as the electronic state of the Fe center may be fine-tuned.
Structure of the NO-treated βY72F mutant
We determined the structure of the NO-treated βY72F mutant without light illumination (Fig. 6). Except for the mutated site and the Fe center, the structure is very similar to that of the wild type, including the hydration water molecules. The average RMSD with the wild type is 0.09 Å for all atoms. The sixth ligand site is occupied not by an NO molecule but by a water molecule, meaning that either the βY72F mutant was not associated with NO or the NO ligand was released during purification in the dark. As suggested by the UV–vis absorption spectral measurement (Fig. 2c), αCys114-SO(H) was found to be oxygenated to Cys-SO2H. Since ReNHase loses its catalytic activity upon the specific oxidation of αCys114-SO(H) to Cys-SO2H [21], we could not interpret whether βTyr72 is involved in the catalytic reaction.

Structure around the catalytic cavity of the βY72F mutant ReNHase. F o–F c omit electron density maps at 3.0σ contour (blue mesh) were superimposed on the refined structures. The sulfinic acid groups of αCys112-SO2 − and αCys114-SO2 − were omitted from the calculations of the F o–F c map. Green, gray, blue, red, yellow, and orange sticks represent carbon (βY72F), carbon (wild type), nitrogen, oxygen, sulfur, and iron atoms, respectively. The coordinated water and hydration water molecules for the βY72F mutant and the wild type are shown as red and brown balls, respectively
Roles of αSer113 and βTyr72 in the catalytic mechanism of NHase
On the basis of detailed kinetic analyses of PtNHase [31, 32] and CtNHase [33], the Ser ligand (αSer113 in ReNHase) was postulated to function as a general base and to ionize the structurally adjacent, conserved Tyr residue (βTyr72 in ReNHase), which activated a water molecule (Fig. 1b). However, this model must be revised because the present study revealed that αSer113 is not directly involved in the catalytic mechanism. Rather, as shown earlier, the hydroxyl group of αSer113 is likely to be important for the electronic state and the structure around the catalytic cavity. The αS113A mutation induced disorder in the side chain of βTyr76 and the two additional hydration water molecules, making distinctly different hydrogen-bond networks in the cavity. These alterations were reversed by the addition of the substrate, the product, or n-BA, meaning that the hydroxyl group of αSer113 is not essential for catalysis.
Unlike the wild-type ReNHase, the βY72F mutant could not be obtained in the nitrosylated state, irrespective of NO treatment. This suggests that the phenol oxygen of βTyr72 affects the electronic state (perhaps the Lewis acidity) of the Fe center in ReNHase to weaken NO binding to the iron. E. coli cells expressing the βY72F mutant ReNHase showed no NHase activity, whereas E. coli cells expressing the wild-type or the αS113A mutant ReNHase showed significant activity. The expression levels of each recombinant enzyme were similar. Considering the reductive environment in living cells, the βY72F mutation might abolish the catalytic activity. In PtNHase, the corresponding Tyr mutant enzyme (βY68F) retained the αCys113-SO(H) modification but showed decreases in K m and k cat by factors of approximately 10 and approximately 100, respectively [32]. The structure of the βY68F mutant PtNHase was found to be almost identical to that of the wild-type enzyme [32]. Although the detailed function of the strictly conserved Tyr residue remains unclear, it is likely to play an important role in the catalysis.
We note that the present study does not contradict the catalytic mechanism in which the sulfenate oxygen of αCys114-SO(H) activates the water molecule making a nucleophilic attack on the nitrile carbon (Fig. 1a). The increased K m and decreased k cat values in the αS113A mutant NHase could be attributed to the significant alteration in the hydrogen-bonding network as well as the electronic state of the Fe center in the substrate-free state. These alterations could disappear when the substrate enters the binding pocket. The αS113A mutation would have little effect on the chemistry of αCys114-SO(H) during the catalytic reaction.
Abbreviations
- CtNHase:
-
Nitrile hydratase from Comamonas testosteroni Ni1
- Cys-SO2H:
-
Cysteine sulfinic acid
- Cys-SOH:
-
Cysteine sulfenic acid
- HPLC:
-
High-performance liquid chromatography
- n-BA:
-
n-Butyric acid
- NHase:
-
Nitrile hydratase
- PDB:
-
Protein Data Bank
- PtNHase:
-
Nitrile hydratase from Pesudonocardia thermophila JCM 3095
- ReNHase:
-
Nitrile hydratase from Rhodococcus erythropolis N771
- RMSD:
-
Root-mean-square deviation
- SCNase:
-
Thiocyanate hydrolase
- t-BuCN:
-
tert-Butylnitrile
- t-BuCONH2:
-
Trimethylacetamide
- t-BuNC:
-
tert-Butylisonitrile
- Tris:
-
Tris(hydroxymethyl)aminomethane
References
Asano Y, Tani Y, Yamada H (1980) Agric Biol Chem 44:2251–2252
Kobayashi M, Shimizu S (1998) Nat Biotechnol 16:733–736
Endo I, Nojiri M, Tsujimura M, Nakasako M, Nagashima S, Yohda M, Odaka M (2001) J Inorg Biochem 83:247–253
Sugiura Y, Kuwahara J, Nagasawa Y, Yamada Y (1987) J Am Chem Soc 154:522–528
Brennan BA, Alms G, Nelson MJ, Durney LT, Scarrow RC (1996) J Am Chem Soc 118:9144–9145
Kopf MA, Bonnet D, Artaud I, Petre D, Mansuy D (1996) Eur J Biochem 240:239–244
Odaka M, Fujii K, Hoshino M, Noguchi T, Tsujimura M, Nagashima S, Yohda M, Nagamune T, Inoue Y, Endo I (1997) J Am Chem Soc 119:3785–3791
Noguchi T, Hoshino M, Tsujimura M, Odaka M, Inoue Y, Endo I (1996) Biochemistry 35:16777–16781
Bonnet D, Artaud I, Moali C, Pétré D, Mansuy D (1997) FEBS Lett 409:216–220
Huang W, Jia J, Cummings J, Nelson M, Schneider G, Lindqvist Y (1997) Structure 5:691–699
Nagashima S, Nakasako M, Dohmae N, Tsujimura M, Takio K, Odaka M, Yohda M, Kamiya N, Endo I (1998) Nat Struct Biol 5:347–351
Miyanaga A, Fushinobu S, Ito K, Wakagi T (2001) Biochem Biophys Res Commun 288:1169–1174
Hourai S, Miki M, Takashima Y, Mitsuda S, Yanagi K (2003) Biochem Biophys Res Commun 312:340–345
Arakawa T, Kawano Y, Kataoka S, Katayama Y, Kamiya N, Yohda M, Odaka M (2007) J Mol Biol 366:1497–1509
Noguchi T, Nojiri M, Takei K, Odaka M, Kamiya N (2003) Biochemistry 42:11642–11650
Dey A, Chow M, Taniguchi K, Lugo-Mas P, Davin S, Maeda M, Kovacs J, Odaka M, Hodgson K, Hedman B, Solomon EI (2006) J Am Chem Soc 128:533–541
Nakasako M, Odaka M, Yohda M, Dohmae N, Takio K, Kamiya N, Endo I (1999) Biochemistry 38:9887–9898
Kovacs JA (2004) Chem Rev 104:825–848
Yano T, Ozawa T, Masuda H (2008) Chem Lett 37:672–677
Murakami T, Nojiri M, Nakayama H, Odaka M, Yohda M, Dohmae N, Takio K, Nagamune T, Endo I (2000) Protein Sci 9:1024–1030
Tsujimura M, Odaka M, Nakayama H, Dohmae N, Koshino H, Asami T, Hoshino M, Takio K, Yoshida S, Maeda M, Endo I (2003) J Am Chem Soc 125:11532–11538
Noveron JC, Olmstead MM, Mascharak PK (1999) J Am Chem Soc 121:3553–3554
Tyler LA, Noveron JC, Olmstead MM, Mascharak PK (2003) Inorg Chem 42:5751–5761
Heinrich L, Mary-Verla A, Li Y, Vassermann J, Chottard JC (2001) Eur J Inorg Chem 9:2203–2206
Lugo-Mas P, Dey A, Xu L, Davin S, Benedict J, Kaminsky W, Hodgson K, Hedman B, Solomon EI, Kovacs J (2006) J Am Chem Soc 128:11211–11221
Yano T, Wasada-Tsutsui Y, Arii H, Yamaguchi S, Funahashi Y, Ozawa T, Masuda H (2007) Inorg Chem 46:10345–10353
Hashimoto K, Suzuki H, Taniguchi K, Noguchi T, Yohda M, Odaka M (2008) J Biol Chem 283:36617–36623
Taniguchi K, Murata K, Murakami Y, Takahashi S, Nakamura T, Hashimoto K, Koshino K, Dohmae N, Yohda M, Hirose T, Maeda M, Odaka M (2008) J Bioeng Biosci 106:174–179
Hopmann KH, Guo JD, Himo F (2008) Inorg Chem 46:4850–4856
Arakawa T, Kawano Y, Katayama Y, Nakayama H, Dohmae N, Yohda M, Odaka M (2009) J Am Chem Soc 131:14838–14843
Mitra S, Holz RC (2007) J Biol Chem 282:7397–7404
Miyanaga A, Fushinobu S, Ito K, Shoun H, Wakagi T (2004) Eur J Biochem 271:429–438
Rao S, Holz RC (2008) Biochemistry 47:12057–12064
Nojiri M, Yohda M, Odaka M, Matsushita Y, Tsujimura M, Yoshida T, Dohmae N, Takio K, Endo I (1999) J Biochem (Tokyo) 125:696–704
Takarada H, Kawano Y, Hashimoto K, Nakayama H, Ueda S, Yohda M, Kamiya N, Dohmae N, Maeda M, Odaka M (2006) Biosci Biotechnol Biochem 70:881–889
Tsujimura M, Odaka M, Nagashima S, Yohda M, Endo I (1996) J Biochem (Tokyo) 119:407–413
White A, Handler P, Smith EL, Hill RL, Lehman IR (1978) In: Principles of biochemistry, 6th edn. McGraw-Hill, New York, pp 196–220
Otowinowski Z, Minor W (1997) Methods Enzymol 276:307–326
Vagin A, Teplyakov A (1997) J Appl Crystallogr 30:1022–1024
CCP4 (Collaborative Computational Project, Number 4) (1994) Acta Crystallogr D Biol Crystallogr 50:760–763
Murshudov GN, Vargin AA, Dodson EJ (1997) Acta Crystallogr Sect D 53:240–255
Sheldrick GM, Schneider TR (1997) Methods Enzymol 277:319–343
Emsley P, Cowtan K (2004) Acta Crystallogr Sect D 60:2126–2132
Read RJ (1986) Acta Crystallogr Sect A 42:140–149
Piersma SR, Nojiri M, Tsujimura M, Noguchi T, Odaka M, Yohda M, Inoue Y, Endo I (2000) J Inorg Biochem 80:283–288
Acknowledgments
We thank the beamline assistants at the Photon Factory for data collection at beamlines NW12A and BL5A. This work was supported in part by a Grant-in-Aid for Scientific Research from the Scientific Research (B) KAKENHI 19350080 (to M.O.) and (B) KAKENHI 21350089 (to M.O.). This work was performed with the approval of the Photon Factory Advisory Committee (approval no. 2008G640).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Yamanaka, Y., Hashimoto, K., Ohtaki, A. et al. Kinetic and structural studies on roles of the serine ligand and a strictly conserved tyrosine residue in nitrile hydratase. J Biol Inorg Chem 15, 655–665 (2010). https://doi.org/10.1007/s00775-010-0632-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-010-0632-3