
Abstract
Copper-transporting ATPases (Cu-ATPases) ATP7A and ATP7B play an essential role in human physiological function. Their primary function is to deliver copper to the secretory pathway and export excess copper from the cell for removal or further utilization. Cells employ Cu-ATPases in numerous physiological processes that include the biosynthesis of copper-dependent enzymes, lactation, and response to hypoxia. Biochemical studies of human Cu-ATPases and their orthologs have demonstrated that Cu-ATPases share many common structural and mechanistic characteristics with other members of the P-type ATPase family. Nevertheless, the Cu-ATPases have a unique coordinate environment for their ligands, copper and ATP, and additional domains that are required for sophisticated regulation of their intracellular localization and activity. Here, we review recent progress that has been made in understanding the structure of Cu-ATPases from the analysis of their individual domains and orthologs from microorganisms, and speculate about the implications of these findings for the function and regulation of human copper pumps.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Intracellular localization determines specific function of copper-transporting ATPase in a cell
Copper-transporting ATPases (Cu-ATPases) represent a large family of membrane transporters found in all phyla. In eukaryotes, the most studied Cu-ATPases are mammalian ATP7A and ATP7B (Menkes disease protein and Wilson disease protein, respectively) [1–3] and yeast Ccc2 [4]. Various bacteria and archaea contain Cu-ATPases, the best characterized of which are CopA and CopB from Bacillus subtilis, Archaeoglobus fulgidus [5, 6], and Thermotoga maritima [7]. Information on other prokaryotic orthologs is also beginning to emerge [8, 9].
It is thought that the Cu-ATPases perform at least three major functions. In some microorganisms, such as Enterococcus hirae and Pseudomonas aeruginosa Q9I147, Cu-ATPases appear to mediate copper uptake [9, 10]. Disruption of the copA gene in E. hirae causes a cessation of cell growth in media containing chelated copper (i.e,. under copper-limiting conditions), whereas wild-type E. hirae continues to grow [9, 10]. More directly, the role of Cu-ATPases in inward copper transport was demonstrated in P. aeruginosa, where cells that express Cu-ATPase HmtA have greater copper uptake than cells that do not [9, 10].
Another function is to maintain intracellular copper levels and prevent toxic copper accumulation. This is achieved either by direct copper extrusion from the cytosol across the plasma membrane (in bacteria and archaea) or by copper sequestration in vesicles/vacuoles (in eukaryotes) for further export or storage. The export/detoxification function is common for Cu-ATPases in all phyla and could have been the first to appear in evolution. An interesting example of copper detoxification is reported for A. fulgidus, which expresses Cu-ATPase CopA. In response to environmental copper elevation, A. fulgidus forms biofilms, heterogeneous structures at the cell surface composed of protein and polysaccharides [10]. These structures also have a very high copper content. It is tempting to speculate that CopA couples its known export function with copper immobilization in biofilms [10]. The inactivation of the Cu-ATPase-driven export of copper and the resultant copper accumulation have deleterious effects on growth and development of all organisms studied so far.
In eukaryotic cells (from yeast to mammals and plants), in addition to metal export, Cu-ATPases participate in the biosynthesis of copper-dependent enzymes in the secretory pathway and in chloroplasts. Copper delivery to the secretory pathway is obligatory for pigmentation, amidation of neuropeptides, normal iron metabolism, inflammatory response, and many other physiological reactions. Disruption of copper transport in eukaryotes has severe pathological multisystemic manifestations, such as the human diseases Menkes and Wilson, and the calamity phenotype seen in zebrafish [11].
The intracellular localization of the Cu-ATPases varies between kingdoms and reflects their primary physiological role(s) in these organisms. In Escherichia coli, CopA is found at the plasma membrane [12], where it performs the detoxification function. The yeast Cu-ATPase, Ccc2, functions in the membrane of the Golgi network [4]. Ccc2 is the major supplier of copper to Fet3, a copper-dependent ferroxidase, and it does not seem to play a significant role in the removal of copper from the cell. Mammalian Cu-ATPases, ATP7A and ATP7B, are found predominantly in the membranes of the trans-Golgi network (TGN) and endocytic vesicles, and to a lesser extent at the plasma membrane [13–15]. ATP7A and ATP7B cycle between these compartments in response to changing copper levels, as well as other stimuli [2, 16]. This regulated intracellular localization determines whether copper is utilized for the biosynthesis of copper-dependent enzymes in the secretory pathway, sequestered in vesicles for storage, or rapidly released from vesicles into the extracellular milieu for export and, possibly, signaling. Finally, in photosynthetic microorganisms and plants, copper is also required in thylakoids, where it is incorporated into plastocyanin and cytochrome oxidase. The photosynthetic organisms have additional Cu-ATPases in thylakoid membranes that are involved in copper delivery to the lumen of this specialized cell compartment [17].
The transport mechanism of Cu-ATPases has distinct features compared with that of other P-type pumps
Cu-ATPases belong to the family of P1B-type ATPases that transport transition metal ions, such as Cu(I), Cu(II), Cd2+, Co2+, Pb2+, and Zn2+. As all P-type ATPases, Cu-ATPases utilize the energy of ATP hydrolysis to translocate metal ion across a lipid bilayer. The key steps in this process involve binding of copper and ATP at the cytosolic portion of the ATPase, transfer of copper to the transmembrane portion of the transporter, hydrolysis of ATP and autophosphorylation, release of copper to the luminal (extracellular) side, and subsequent dephosphorylation that resets the cycle. This general transport mechanism also exists in other members of the P-type ATPase family. However, specific details of substrate recognition, binding affinities, and the dependence of activity on the presence of reducing agents are unique for Cu-ATPases. Current data indicate that whereas the Cu-ATPases have domain architecture similar to that of other P-type pumps, they also have characteristic domains and sequence motifs that are only found in the P1B subfamily (see later for details). Common features of Cu-ATPases across species have been targets of study in understanding the copper transport mechanism and the ligand specificity of these proteins. It is now clear that sequence motifs common to P1B-type ATPases that are not present in other P-type ATPases are involved in nucleotide recognition and metal selectivity.
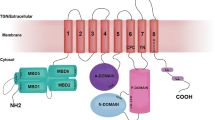
P-type ATPases use the energy released upon hydrolysis of ATP to drive the translocation of ions across a membrane. Like other P-type ATPases, Cu-ATPases hydrolyze ATP with the formation of a transient phosphorylated intermediate. Unlike the better studied P2-type ATPases (Ca2+-ATPase, Na+,K+-ATPase) or P3-type H+-ATPase, human Cu-ATPases do not generate a gradient of transported ion. In eukaryotic cells, Cu-ATPases also do not operate against an existing ion gradient, because all copper inside and outside the cell is thought to be bound to various proteins and, possibly, other cell constituents. Rather, eukaryotic Cu-ATPases retrieve copper from copper carriers (copper chaperones) and then transfer it across the membrane for incorporation into acceptor molecules (Fig. 1).

General organization of human copper-transporting ATPases (Cu-ATPases). Cu-ATPases have three main functional units. The transmembrane domain provides a translocation pathway and has a copper binding site or copper binding sites essential for the catalytic and transport activity. The ATP hydrolysis is confined to the cytosolic portion and is mediated through cooperation of three structural domains (see the text for details). The N-terminal domain (with six copper-binding subdomains) senses changing copper levels and regulates the Cu-ATPase activity. The transport cycle is initiated by the transfer of copper from intracellular donor Atox1 and may be facilitated by apo acceptor molecules
Thus, the metallation state of copper donors (chaperones) and acceptors is likely to play a more important role in determining the rate of copper transport by ATP7A and ATP7B than the total copper content in a cell. The effect of the copper chaperone Atox1 on the activity of human Cu-ATPases has been directly demonstrated [18]. In the copper-bound form, Atox1 stimulates the catalytic activity of ATP7B, whereas the apo form of Atox1 decreases the activity of ATP7B, presumably by removing copper from the regulatory sites (see later). Similarly, the archaeal copper donor metallochaperone CopZ, in the copper-bound form stimulates the ATPase activity of CopA [19]. Whether and how copper acceptors modulate the transport activity of Cu-ATPases remains to be determined.
The difference in primary physiological functions of mammalian P2-type ATPases, P3-type ATPases, and Cu-ATPases (the generation and maintenance of an ion gradient as opposed to the biosynthesis of copper-dependent enzymes) may explain a very significant (about 2 orders of magnitude [20]) difference of their turnover, i.e., Cu-ATPases appear to be much slower compared with other pumps. Recent studies yielded the first quantitative estimates of Cu-ATPase activity. The recombinant Cu-ATPase CopA from two different thermophiles has been purified, and the enzymatic properties of this protein have now been studied in significant detail, although transport has not yet been reconstituted [7, 21–23]. These studies revealed catalytic activity of T. maritima CopA with a V max of 1.8–2.7 μmol/mg protein/min [23] and 1.3 μmol/mg protein/min for A. fulgidus CopA [22]. The values for T. maritima CopA activity likely do not represent the enzyme maximum activity because measurements were carried out at 60 °C, whereas the optimum temperature for Thermatoga growth is 70–80 °C [24].
Only one report on the purification of eukaryotic Cu-ATPases has been published to date [25]. In this study, the copper-dependent ATPase activity of purified ATP7A was not reported, but transport measurements revealed a rate of 44.9 pmol Cu/mg protein/min. More recent experiments using adenovirus-mediated expression of ATP7B in mammalian cells yielded the first measurements of the ATPase activity for ATP7B in native membranes [20]. The value of 30 nmol/mg protein/min obtained is for the microsomal protein (where ATP7B is 10–20% of the total), i.e., the activity of purified ATP7B is expected to be significantly higher. One should note that in this latter report, ATP7B was highly overexpressed and the intracellular localization in the TGN was not strictly maintained. Thus, the reported activity represents an average of specific activities of ATP7B in different cell compartments. Presently, it is unknown whether ATP7A and ATP7B have different turnover rates depending on their intracellular localization. However, both proteins undergo regulatory kinase-mediated phosphorylation that is different for the Cu-ATPases in the TGN and vesicles [26, 27]. Therefore, it is conceivable that the transport activity of ATP7A and ATP7B can also be regulated when they traffic from one cell compartment to the other. In fact, preincubation of microsomal membranes with phosphatase increases the rate of phosphorylation in vitro [28].
The rate-limiting step of the catalytic cycle of Cu-ATPases is unknown. By analogy with other P-type pumps, the conformational transition that changes the orientation of the intramembrane sites allowing for ion release could be rate-limiting. However, it is possible that the rates of other steps may significantly influence overall turnover. For example, the removal of copper from the cytosolic copper binding sites using a copper chelator has an unexpected retardation effect on the rate of dephosphorylation of CopA [23], suggesting that constant occupation of the N-terminal regulatory sites by copper could be essential for enzyme turnover.
The copper chaperone CopZ for A. fulgidus CopA is unique in that it has an additional [2Fe–2S]-containing domain, which can reduce Cu(II) to Cu(I). This observation led to the fascinating possibility that the transport of copper by A. fulgidus CopA is a coupled process, involving metal reduction as its first step [29]. Whereas the structure of A. fulgidus CopZ is unique, the requirement for the presence of a reducing agent during transport is common for Cu-ATPases [19, 30, 31]. This could be a simple reflection of a need to keep copper in the reduced form. However, the fact that ascorbate (which reduces copper) cannot substitute for the presence of sulfhydryl-reducing reagents (such as dithiothreitol or glutathione) points to an interesting possibility that the reduced state of both protein and copper is essential for transport. In CopA, in addition to reducing agents, the presence of millimolar concentration of Cys upregulates the enzyme’s turnover [32], although the mechanism of this stimulation is not entirely clear.
Overall, recent progress in the characterization of the functional properties of Cu-ATPases has revealed many interesting and distinct aspects of their transport mechanism. In particular, archaeal and bacterial pumps are emerging as excellent models for characterization of the structure and biochemistry of Cu-ATPases. At the same time, certain caution is needed when generalizations are made. Cell physiological processes and environmental conditions are markedly different for sulfur-metabolizing archaea or bacteria grown in thermal vents, mesophilic bacteria, and eukaryotic/mammalian transporters. For example, hydrogen inhibits the growth of T. maritima, but inhibition can be overcome by the addition of sulfur, which forms H2S. Thus, it seems unlikely that a high concentration of protons would have a stimulatory effect on copper transport activity of T. maritima CopA. By contrast, lowering the pH was reported to increase the transport activity of human ATP7B [33]. A sharp pH dependence with a maximum around pH 6.25 was reported for CopB from E. hirae [34]. Complementary analysis of Cu-ATPases from different species that links their functional diversity to specific physiological environments in which they operate promises to greatly enrich our understanding of copper metabolism across species.
The molecular design of Cu-ATPases reflects both common and unique aspects of their transport mechanism
P-type ATPases are transmembrane proteins that have a common core structure (Fig. 2). The core structure includes cytosolic domains for autocatalytic phosphorylation (P-domain), nucleotide binding (N-domain), and energy transduction (responsible for the structural transfer of free energy) (A-domain).

The A and P cytosolic domains are directly linked to the transmembrane segments; the N-domain is an insertion within the P-domain. The N- and C-termini, both oriented in the cytosol, vary in length, and often contain additional regulatory domains or motifs. The Cu-ATPases have a distinct N-terminal metal-binding domain (MBD), composed of one to six structurally similar subdomains (Fig. 1). The C-terminus of Cu-ATPases may contain an additional metal binding site (as in archaeal CopA) or determinants for endocytosis and kinase-mediated phosphorylation (as in mammalian transporters).
Unlike other members of the P-type ATPases, which contain ten transmembrane helices, the Cu-ATPases have eight transmembrane helices (TM1–TM8). The transmembrane sequences are not highly conserved within the Cu-ATPase subfamily, with the notable exception of TM6–TM8. These segments contain the CPC/H, NxxxxxxYN, and MxxSS/TxxV motifs, respectively. The first two motifs are invariant in all ATPases shown or predicted to be involved in copper transport [37] and the last motif is highly conserved. The residues in all three motifs are thought to contribute to the intramembrane copper binding sites and participate in copper translocation across the membrane.
Topologically, TM5–TM8 closely match TM3–TM6 of the Ca2+-ATPase (SERCA1), Na+,K+-ATPase, or H+-ATPase, for which high-resolution structures have been determined and involvement in ion coordination has been directly demonstrated. It is thought that along with the cytoplasmic domains of the protein, TM4–TM8 form part of a conserved functional core found in all P-type ATPases [38, 39]. The spatial location of TM1–TM3 in the bundle is less clear owing to the lack of homology to better characterized ATPases. One possible location of all transmembrane helices for E. hirae CopA has been predicted by Lubben et al. [40]; this model places the TM6 and TM7 CPC and YN motifs directly facing one another (Fig. 3).

Predicted orientation of transmembrane (TM) helices [40]. CPC motif in yellow, YN motif in pink
The ATP-binding domain of Cu-ATPases binds ATP at residues unique to the P1B-type ATPase family
The ATP-binding domain of Cu-ATPases is highly conserved from bacteria to humans (Fig. 4). Cu-ATPases, as all P-type ATPases, contain conserved sequence motif D-K-T-G-T-[LIVM]-[TIS] in the P-domain (Fig. 2, insert). The invariant Asp in this motif is central for their catalytic activity. During ATP hydrolysis, the Asp accepts γ-phosphate from ATP bound in the N-domain (Fig. 2b) and becomes reversibly phosphorylated with the formation of an acylphosphate intermediate. It is subsequently dephosphorylated by an intrinsic phosphatase activity as part of the catalytic cycle during cation transport [38, 41–43].

Nucleotide-binding-domain alignment of human proteins ATP7A and ATP7B with bacterial and archaeal CopA. Identical residues are in blue and conserved residues within two or more sequences are in gray. The Conserved SEHP domain is shown in pink. B. sub, Bacillus subtilis; A. ful Archaeoglobus fulgidus
The N- and P-domains provide an interesting example of evolution at work in the P-type ATPase family. Although the sequence and structure of the P-domain portion of the ATP-binding domain are highly similar for all characterized P-type ATPases, including Cu-ATPases, the primary sequence of the N-domain is markedly different in Cu-ATPases compared with other P-type pumps. Structurally, the P-domain is a member of the haloacid dehalogenase family, which consists largely of small soluble proteins, such as phosphatases, epoxide hydrolases, and l-2-haloacid dehalogenases (HAD) [44]. Analysis of this family illustrates that the common “P-domain-like structures” mediate catalysis, whereas the delivery of various substrates to the active site is done by the structurally unrelated inserts into the P-domain. (In the case of P-type ATPases, it is the independently folded N-domain that delivers and positions ATP in such a way that the transfer of phosphate to the invariant Asp can occur.) It is interesting to consider that many members of the HAD superfamily are involved in detoxification of xenobiotics or metabolic by-products [44] and that the original function of Cu-ATPases was likely the removal of excess copper, whereas the role in cofactor delivery evolved later.
In contrast to the P-domain of Cu-ATPases, which shares its sequence and fold with all P-type ATPases, the sequence of the N-domain is only conserved between the members of P1B family. Nevertheless, despite the lack of sequence conservation with other P-type pumps, the fold of the N-domain is conserved, perhaps as an example of evolutionary convergence of structures. The N-domain consists of six antiparallel β-sheets with four α-helices. In ATP7B, five residues were found to be important in nucleotide binding, His1069, Gly1099, Gly1101, Gly1149, and Asn1150, conserved in all Cu-ATPases, and show a significant chemical shift upon binding of nucleotide. The mutagenesis of some of these residues was shown to affect ATP binding [45–47]. The equivalent residues in the ATP-binding domain of CopA (Glu457, His462, and Gly492) were proposed to orient and bind the adenine ring through hydrogen bonding, and Gly490 and Gly501 were thought to interact primarily with the ribose moiety and α-phosphate. Recently, the ATP-binding domain of CopA with bound adenosine 5′-[β,γ- methylene]triphosphate or ADP and Mg2+ was crystallized, confirming the importance of all these residues. The structure also clarified the role of invariant Glu457 in the E457xxSEHPL sequence (Fig. 4), which was shown to form hydrogen bonds with the N1 and N6 atoms of the adenine ring [36].
A-domain and conformational transitions
Another domain common for Cu-ATPases and other P-type pumps is the A-domain (Figs. 2, 5). This domain contains a highly conserved T/SGE motif, which inserts in the cleft between the N- and P-domains during ATP hydrolysis. Mutational [48] and structural [49] studies in Ca2+-ATPase SERCA revealed the role for invariant Glu residues in this motif in activating the water molecule during dephosphorylation. In addition, the entire A-domain rotates and by triggering movements of transmembrane helices regulates the release of the ion at the luminal site [49, 50]. Recent studies on bacterial CopA have indicated that similar movements occur in copper pumps [39]. However, the structural basis of interdomain interactions is likely to differ. For example, in Ca2+-ATPase, the side chain of the Ser residue in the TGES sequence forms a hydrogen bond with the invariant Asp residue in the TGDN sequence at the N-domain/P-domain interface. In Cu-ATPases, the Ser residue is substituted with Ala or Pro, and thus the nature of the interaction between the two motifs cannot be the same.

The A-domain structure and rotation during catalysis. An overlay of A-domain structures for SERCA (blue) and CopA (green) is shown on the left. The location of the RADRK loop of SERCA (yellow) in the ATP-bound state (middle) and following phosphorylation with magnesium phosphate (right)
An overlay of available A-domain structures for Ca2+ and Cu-ATPases (Fig. 5, left) also illustrates that despite a very similar fold of the core of the domain, the length and the orientation of the loops differ considerably. The difference in the size and the orientation of the loops may become important if one considers their location in the overall structure. For example, the TLGEDNLI loop of ATP7B has a position equivalent to that of the RADRK sequence in SERCA. In SERCA, the exposure of this loop changes dramatically when the A-domain rotates (Fig. 5, middle and right). In Cu-ATPases, the A-domain undergoes apparently similar rotation as evidenced by a marked change in proteolytic susceptibility of regions connecting the A-domain to the transmembrane portion of the protein [39]. Thus, it is conceivable that the equivalent loop will become similarly exposed when protein is phosphorylated and can serve as a target of interaction with regulatory domains and/or other proteins. It could be coincidental, but nevertheless intriguing, that the replacement of the TGE motif with the AAA sequence in human Cu-ATPases is associated with protein hyperphosphorylation and constitutive trafficking from the TGN to vesicles [42].
Structure and arrangement of cytosolic MBDs
All Cu-ATPases have at least one conserved copper-binding motif, MxCxxC, in the N-terminal tail, with the number of these domains varying between species. Human ATP7A and ATP7B have six copper binding sites in the N-terminal copper-binding domain [51, 52]. The structures of all six subdomains in the N-terminus of ATP7A have been determined by NMR and crystallography (Protein Data Bank accession numbers 1KVJ, 1S6U, 2GA7, 2AW0, 1Y3J, and 1YJV) in both copper-loaded and apo forms. In addition, the structures of two pairs, MBD5/MBD6 and MBD3/MBD4, have been characterized for ATP7B [53, 54]. The structure of individual MBDs revealed a ferredoxin-like fold in each domain, and the structures are largely superimposable (Fig. 6, left).

The structure of and copper binding by individual N-terminal metal-binding domains (MBDs). Left Overlay of MBD2, MBD4, and MBD6 of ATP7A. Middle Copper (red ball) is coordinated in the exposed loop GMXCXXC by two Cys residues (yellow). Right Copper binding does not affect the structure of the domain but alters the orientation and dynamics of the loops. An overlay of apo (yellow) and copper-bound (green) MBD2 of ATP7A is shown; GMTCxxC is in red
X-ray absorption spectroscopy of MBD2 of ATP7A [55] and ATP7B [56] showed that copper is coordinated with a linear biscysteinate geometry with the CxxC motif located on an exposed loop of the protein (Fig. 6, middle). Upon copper binding in constructs containing only one or two domains, the ferredoxin fold of the MBD remains unaltered (Fig. 6, right), with structural changes mainly affecting the CxxC-containing loop and the N-terminus of the first α-helix in the domain [53–55, 57–60].
In contrast to the individual MBDs, copper binding to the full-length N-terminal domain of ATP7B, which includes all six MBDs, induces significant changes in protein structure detectable by circular dichroism spectroscopy [61]. This observation indicates that in the full-length N-terminus, small copper-induced changes in the dynamics and orientation of the loops within or between the individual MBDs are propagated and amplified, causing larger changes in the entire protein structure. These conformational changes have important consequences for protein function: they affect interdomain interactions, allow access to interacting proteins, and provide sites for kinase-mediated phosphorylation (see later for details). How does this communication between MBDs occur? A hydrogen-bonding network may play an important role in holding individual domains together. The presence of such a network has been experimentally demonstrated for the MBD5/MBD6 pair [54]. In this pair, individual MBDs are connected by a very short linker, and the entire pair forms a rigid-body structure that is likely to move in unison.
The rigid-body structure of the MBD5/MBD6 pair provides an explanation for the earlier puzzling results in site-directed mutagenesis studies of the N-terminal domain of ATP7A and ATP7B. Specifically, it was shown that the inactivation of copper binding in both MBD5 and MBD6 is deleterious for the transport activity of ATP7A and ATP7B, whereas Cu-ATPase containing the metal binding site in one of these MBDs is functional [62–64]. It is tempting to speculate that MBD5/MBD6 mediates its activity through interaction with other functional domains (see later) and that the binding of copper to either site induces movement of the entire MBD5/MBD6 structure.
Although rigid-body movement is an appealing hypothesis, some data in the literature appear to disagree with the idea of fixed mutual orientations of MBDs. For example, the mutual orientation of MBD5/MBD6 of ATP7B differs significantly from the orientation of the presumably functionally equivalent MBD1/MBD2 pair of CopA from B. subtilis (Fig. 7). This observation may reflect either artificial packing during crystallization or, more interestingly, the ability of the MBDs to rotate with respect to each other during transport (perhaps following the rotation of the A-domain, with which MBDs interact).

Orientation of two MBDs connected by a short linker in ATP7B (purple; MBD5/MBD6 Protein Data Bank accession number 2EWP) and B. subtilis CopA (yellow, MBD1/MBD2, Protein Data Bank accession number 1P6T). The first metal-binding subdomains in each pair are aligned
Whereas the MBDs closest to the membrane are connected by a very short linker, most of the N-terminal MBDs in human pumps are linked by long loops. Structural analyses of the MBD1–MBD6 region led to the suggestion that all subdomains act independently [65, 66], and a similar conclusion was reached for the MBD3/MBD4 pair of ATP7B [53]. However, the refolding protocol used for protein purification in these studies may have affected the structure of the loops and disrupted the interdomain contacts. The findings of functional studies on protein purified without refolding indicate that copper binding to one MBD alters the loop structure and availability of other MBDs [28, 56], i.e., MBDs communicate. Furthermore, X-ray spectroscopic analysis of MBD1–MBD6 fully loaded with copper [67] indicates that some metal binding sites are within close proximity to one another, resulting in a Cu–Cu spectroscopic signal. Because the structure of MBD5/MBD6 positions the metal binding sites at opposite ends where copper atoms would not interact, one must conclude that either different MBD pairs within the full-size protein come together head-to-tail or long loops connecting the MBDs within the MBD1–MBD4 region allow for an orientation in which metal binding sites approach one another (Fig. 8).

Predicted orientation of two N-terminal MBDs connected by a long loop. The modeling for the MBD1 (purple) to MBD2 (green) protein region was done ab initio using the Rosetta software suite. The metal-binding sequence GMTCxxC in each MBD is shown in yellow
Copper transfer from a cytosolic chaperone to Cu-ATPase
Copper does not bind the Cu-ATPases as a free ion, but is transferred from a class of proteins known as metallochaperones. The most studied metallochaperones include CopZ, which transfers copper to the bacterial and archaeal Cu-ATPases, Atox1 or HAH1 found in mammalian systems, and Atx1 in yeast [68–71]. Whereas bacterial CopZ, Atox1, and Atx1 show a protein fold similarity to the MBDs of the Cu-ATPases [72], archaeal CopZ and similar metallochaperones from plants often contain species-specific domains, in addition to an Atx1-like domain, that may serve a species-specific regulatory function [29, 73, 74] (see earlier). Like the Cu-ATPases, copper chaperones contain an MxCxxC copper-binding sequence [70, 75] and bind copper through two sulfurs [76, 77]. In human Cu-ATPases, the K a for copper does not differ significantly for the chaperone and Cu-ATPase MBDs, but MBDs retain copper better [66], favoring vectorial transfer rather than equilibrium.
The recombinant N-terminal MBDs of ATP7B and ATP7A can be reconstituted completely in vitro with Cu(I)–acetonitrile or in cells by growing E. coli cells in a high-copper medium [78, 79]. The in vivo copper donor Atox1 also transfers copper to all six sites in the truncated protein [18, 65]. However, it forms a detectable Cu(I)-mediated complex only with MBD1 and MBD4 of ATP7A [80, 81] and MBD1, MBD2, and MBD4 of ATP7B [65]. This observation implies that MBD5 and MBD6, which are required for metal transport across the membrane, may receive copper from other MBDs, rather than through direct transfer from the copper chaperone. Indeed, the possibility of copper transfer between the MBDs has been shown for MBD 1 and MBD4, and MBD4 and MBD5 [54, 82]. However, mouse ATP7B lacks functional MBD4 [61], thereby complicating a hypothesis involving copper transfer from MBD4 to other MBDs.
The transfer of copper from Atox1 to the N-terminal domain of the mammalian Cu-ATPases requires specific protein interactions resulting in the formation of chaperone copper–MBD adducts [66, 83, 84]. Labeling studies in isolated N-terminal domain from ATP7B examined the discriminate interactions of the metallochaperone with the N-terminal metal binding sites of ATP7B and found that the transfer of one copper results in the selective protection of the Cys residues of MBD2 [56]. Since it has been established that Atox1 is capable of transferring copper to all of the MBDs, the selective labeling of MBD2 indicates that other domains may be protected from Atox1 interaction by protein folding in the full-length N-terminal domain and that Atox1 gains access to these other MBDs only after MBD2 has been loaded with copper. In a more detailed study of the ATP7A MBD1–Cu–Atox1 adduct, it was recently shown that the interaction involves one Cys residue from Atox1 [85].
Recently, molecular dynamics and quantum mechanical simulations on the Atox1–ATP7A MBD4 model showed that either a two--coordinate or a three-coordinate Cu(I) species is preferred over a four-coordinate intermediate between Atox1 and its target protein [86]. Similarly, in yeast, a three-coordinate intermediate is formed in the transfer of Cu(I) from Atx1 to the first MBD of Ccc2, with Cu(I) bound between the two Cys residues of MBD1 of Ccc2 and one Cys of Atx1 [87]. Chemical-shift analysis of a construct containing only the two metal binding sites in the N-terminal domain of B. subtilis CopA demonstrated that the two Cu(I)-binding motifs exchange copper and form a copper-cluster-containing dimer when copper ratios are above 1 [88]. The formation of a copper cluster would imply that this copper is not available in the delivery of copper to transmembrane sites. In this case, copper binding and cluster formation may induce other structural changes that allow access to the transmembrane copper binding site perhaps with direct transfer from the copper chaperone.
Consistent with this latter mechanism, metal from the N-terminal domain of CopA appeared not to be transferred to the transmembrane site. In A. fulgidus CopA, copper-loaded MBDs are unable to donate copper to the transmembrane copper binding sites of apo-CopA or a truncated CopA missing the N-terminal domain [19]. However, the copper chaperone for this protein, CopZ, transfers copper to both of these constructs.
The transmembrane metal binding sites
Although the overall structure of Cu-ATPases resembles that of other P-type ATPses, the former contain unique copper-binding motifs within the transmembrane portion. Recent studies of archeael CopA revealed the presence of two bound copper atoms (presumably bound within the transmembrane portion) when cytosolic copper-binding domains are deleted. The A. fulgidus CopA has a signature CPC motif in TM6 and requires several other residues for copper binding and translocation, including Tyr682, Asn683, Met711, and Ser715 36. These residues are conserved in the structure of eukaryotic Cu-ATPases, including human ATP7A and ATP7B, and are thought to be copper-coordinating ligands. X-ray spectroscopy and mutagenesis of CopA showed two high-affinity copper binding sites (K a= 1.3 fM−1 and K a = 1.1 fM−1) in the transmembrane region, both with trigonal coordination [89]. It was suggested that one site is formed with the CPC motif in TM6 and a Tyr from TM7. The second copper binding site is made of Asn in TM7 and Met and Ser in TM8. Mutation of corresponding residues decreases the copper-binding stoichiometry, further confirming the importance of these residues in copper binding. Although replacement of the transmembrane Cys residues by Ala in the CPC motif abolishes copper transport and ATP hydrolysis, this enzyme is still able to bind ATP, suggesting that enzyme turnover is prevented by the lack of metal binding [90].
The overall sequence similarity between bacterial CopA and the mammalian transporters ATP7A and ATP7, is approximately 40%, and the copper-binding residues found within the transmembrane domain of CopA are conserved in ATP7A and ATP7B. Owing to this sequence similarity, it is assumed that two copper binding sites are also present in the eukaryotic Cu-ATPases. If reported high-affinity copper binding in the transmembrane domain is common to all Cu-ATPases, it could not only imply the prevention of the release of copper back into the cytosol, but could also indicate that copper release in the lumen is not a diffuse process. In mammalian cells, where copper is transported from Cu-ATPases to target proteins, specific protein–protein interactions may be required to remove copper from the transmembrane sites. Also, a significant rearrangement of the transmembrane portion may be needed to destroy the trigonal coordination environment and greatly decrease the affinity for copper, allowing it to dissociate. Lastly, in the absence of data for transport stoichiometry, it is not clear whether two coppers are released simultaneously or sequentially and how many coppers are actually released per ATP hydrolyzed.
Copper binding to the cytosolic N-terminal domain regulates Cu-ATPase
Current evidence suggests that the role of cytosolic N-terminal domains in Cu-ATPases is regulatory. Studies of E. coli CopA showed that, although cells expressing the Cu-ATPase with its N-terminal domain deleted lose resistance to copper (i.e., copper transport/export function is lost), mutation of the MBD Cys residues has no apparent effect [12, 91]. These studies indicate that the structural presence of the N-terminal domain in E. coli Cu-ATPase is required for copper transport, whereas the metal-binding properties of this domain are not. Replacement of Cys with Ala in the N-terminal MxCxxC domain of A. fulgidus CopA reduces the dephosphorylation rate of the enzyme, even though the mutant binds copper and ATP with the same affinity as wild-type CopA [90]. Deletion of the N-terminal domain, by contrast, had no effect on dephosphorylation but abolished regulation (inhibition) by high levels of copper [22]. These observations illustrate that the consequences of deletion and mutations in different Cu-ATPases are not identical and generalization with respect to the mechanism should be made with caution. Altogether, it appears that the presence of the N-terminal domain may play an important role in resetting the transporter, allowing ATP binding and the initiation of a new cycle. Indeed, when the entire N-terminal copper-binding domain of T. maritima CopA is deleted, negligible ATPase turnover is sustained even though high levels of phosphoenzyme are obtained [23].
Most of the N-terminal binding domain of both ATP7A and ATP7B is not required for copper transport [62, 63, 92]. The presence of only MBD5 or MBD6 is sufficient. Although an adduct was not detected between the copper chaperone and these metal binding sites by NMR, transfer between Atox1 and these domains still occurs [81]. By contrast to CopA, the deletion of the entire N-terminus disrupts the transport function of mammalian pumps. In a yeast complementation study, substitution of Cys for Ser in MBD4–MBD6, MBD5–MBD6, or all six domains resulted in inactivation of the protein and abolished copper-induced trafficking [63]. The inhibitory effect of the deletion is not due to a negative effect on protein folding, because ATP7B with a deleted N-terminus can be produced in quantities similar to those in the wild-type protein and can be delivered to the membrane in the correct orientation [93].
Given an observation that the loss of copper binding to the N-terminal domain inhibits dephosphorylation [90], it is tempting to speculate that the N-terminal domain controls the enzyme transport rate by blocking ATP binding when copper is not present. This effect is mediated via a metal-dependent interaction with the ATP-binding domain (an interaction with the A-domain is likely but has not been explored yet). The copper-bound N-terminal domain of ATP7B associates with the ATP-binding domain less tightly than the apo N-terminal domain [94]. As the interaction between the N-terminal MBD and the ATP-binding domain decreases, the affinity of the ATP-binding domain for ATP increases [95].
Analysis of CopA suggests that like ATP7A and ATP7B, in the copper-free state, the N-terminal MBD may have an autoinhibitory role by interacting with the nucleotide-binding domain and preventing either ATP binding or domain movement necessary for catalytic phosphorylation [35]. Comparison of densities of the EM structure of CopA with and without an N-terminal domain allowed the determination of the position of this domain in the full-length protein (Fig. 2). It is particularly interesting that in this model the N-terminal MBD is strategically positioned between two key functional domains (the N-domain and the A-domain) and that the copper-binding residues in MxCxxC are those closest in proximity to the ATP-binding domain [22], approaching the conserved sequence SEHP (Fig. 9), common to all Cu-ATPases (Fig. 4). Given the proven role of the invariant His in regulating the affinity for ATP, it is easy to imagine how copper binding in proximity to this residue may alter its orientation and influence ATP binding.

Position of the metal-binding motif with respect to the nucleotide-binding domain in the CopA model [22]. Gray N-terminal domain, green ATP-binding domain, orange ExxSEHP motif (His is shown by sticks), red Cys17 and Cys20 in the metal-binding motif MxCxxC
More recently, the N-terminal domain was shown to be a target of kinase-mediated phosphorylation within the MBD3–MDB4 region [28] and the MBD4–MBD5 loop [20]. Phosphorylation of serine residues has been proposed to be an important step in copper homeostasis, allowing for protein–protein interactions conducive to copper transport and trafficking [96]. Analysis by mass spectrometry of proteolysis fragments after copper binding to the N-terminal binding domain of ATP7B shows a structural rearrangement upon copper binding that alters MBD loop exposure, allowing target residues to be phosphorylated by a kinase, leading to intracellular trafficking and/or a functional conformation favorable for copper transport [28]. The latter has been shown to occur in yeast copper transport, where cyclic-AMP-dependent protein kinase phosphorylates Ser258 of Ccc2 and facilitates the energy interconversion step of copper transport [97].
Conclusion
Significant progress has been made in dissecting the structural organization of different functional modules of Cu-ATPases. Studies of bacterial and archaeal orthologs have been particularly helpful in identifying the commonalities and differences in the design and function of copper pumps compared with other P-type ATPases. The functional work led to a better understanding of the unique features of the transport mechanism of Cu-ATPases, such as frequent dependence on the presence of reducing or SH-containing reagents, reliance on copper donors as a part of the transport cycle, unique coordination environment for the binding of copper and ATP, and distinct regulatory role for the N-terminus. We still lack details of many aspects of the structure and function of human Cu-ATPases. Determining the structure for the full-length protein promises to provide information on the arrangement of the copper-translocation pathway, for which information is currently very limited. Understanding the mutual orientation of multiple copper-binding domains in human ATP7A and ATP7B and their changes upon copper binding will provide insights into the mechanisms behind many regulatory effects on copper. It is already apparent that significant conformational changes occur during the Cu-ATPase cycle. Learning about structural elements that become hidden and exposed during these conformational changes is likely to help in identifying interacting proteins and their mechanism of action on human copper pumps.
References
Mercer JF, Llanos RM (2003) Molecular and cellular aspects of copper transport in developing mammals. J Nutr 133:1481S–1484S
Veldhuis NA, Gaeth AP, Pearson RB, Gabriel K, Camakaris J (2009) The multi-layered regulation of copper translocating P-type ATPases. Biometals 22:177–190
Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY (2007) Function and regulation of human copper-transporting ATPases. Physiol Rev 87:1011–1046
Yuan DS, Dancis A, Klausner RD (1997) Restriction of copper export in Saccharomyces cerevisiae to a late Golgi or post-Golgi compartment in the secretory pathway. J Biol Chem 272:25787–25793
Mandal AK, Cheung WD, Arguello JM (2002) Characterization of a thermophilic P-type Ag+/Cu+-ATPase from the extremophile Archaeoglobus fulgidus. J Biol Chem 277:7201–7208
Gaballa A, Helmann JD (2003) Bacillus subtilis CPx-type ATPases: characterization of Cd, Zn, Co and Cu efflux systems. Biometals 16:497–505
Hatori Y, Lewis D, Toyoshima C, Inesi G (2009) Reaction cycle of Thermotoga maritima copper ATPase and conformational characterization of catalytically deficient mutants. Biochemistry 48:4871–4880
Chintalapati S, Al Kurdi R, van Scheltinga AC, Kuhlbrandt W (2008) Membrane structure of CtrA3, a copper-transporting P-type-ATPase from Aquifex aeolicus. J Mol Biol 378:581–595
Lewinson O, Lee AT, Rees DCA (2009) P-type ATPase importer that discriminates between essential and toxic transition metals. Proc Natl Acad Sci USA 106:4677–4682
Odermatt A, Suter H, Krapf R, Solioz M (1993) Primary structure of two P-type ATPases involved in copper homeostasis in Enterococcus hirae. J Biol Chem 268:12775–12779
Mendelsohn BA et al (2006) Atp7a determines a hierarchy of copper metabolism essential for notochord development. Cell Metab 4:155–162
Fan B, Rosen BP (2002) Biochemical characterization of CopA, the Escherichia coli Cu(I)-translocating P-type ATPase. J Biol Chem 277:46987–46992
Petris MJ et al (1996) Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J 15:6084–6095
Suzuki M, Gitlin JD (1999) Intracellular localization of the Menkes and Wilson’s disease proteins and their role in intracellular copper transport. Pediatr Int 41:436–442
Guo Y, Nyasae L, Braiterman LT, Hubbard AL (2005) NH2-terminal signals in ATP7B Cu-ATPase mediate its Cu-dependent anterograde traffic in polarized hepatic cells. Am J Physiol Gastrointest Liver Physiol 289:G904–G916
La Fontaine S, Mercer JF (2007) Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch Biochem Biophys 463:149–167
Abdel-Ghany SE, Muller-Moule P, Niyogi KK, Pilon M, Shikanai T (2005) Two P-type ATPases are required for copper delivery in Arabidopsis thaliana chloroplasts. Plant Cell 17:1233–1251
Walker JM, Tsivkovskii R, Lutsenko S (2002) Metallochaperone Atox1 transfers copper to the NH2-terminal domain of the Wilson’s disease protein and regulates its catalytic activity. J Biol Chem 277:27953–27959
Gonzalez-Guerrero M, Arguello JM (2008) Mechanism of Cu+-transporting ATPases: soluble Cu+ chaperones directly transfer Cu+ to transmembrane transport sites. Proc Natl Acad Sci USA 105:5992–5997
Pilankatta R, Lewis D, Adam CM, Inesi G (2009) High yield heterologous expression of WT and mutant Cu+ ATPase (ATP7B, Wilson disease protein) for functional characterization of catalytic activity and serine residues undergoing copper dependent phosphorylation. J Biol Chem 284:21307–21316
Arguello JM, Mandal AK, Mana-Capelli S (2003) Heavy metal transport CPx-ATPases from the thermophile Archaeoglobus fulgidus. Ann N Y Acad Sci 986:212–218
Rice WJ, Kovalishin A, Stokes DL (2006) Role of metal-binding domains of the copper pump from Archaeoglobus fulgidus. Biochem Biophys Res Commun 348:124–131
Hatori Y et al (2008) Intermediate phosphorylation reactions in the mechanism of ATP utilization by the copper ATPase (CopA) of Thermotoga maritima. J Biol Chem 283:22541–22549
Hensyl WR (ed) (2000) Bergey's manual of determinative bacteriology, 9th edn. Lippincott Williams and Wilkins, Philadelphia, p 304
Hung YH, Layton MJ, Voskoboinik I, Mercer JF, Camakaris J (2007) Purification and membrane reconstitution of catalytically active Menkes copper-transporting P-type ATPase (MNK; ATP7A). Biochem J 401:569–579
Vanderwerf SM, Cooper MJ, Stetsenko IV, Lutsenko S (2001) Copper specifically regulates intracellular phosphorylation of the Wilson’s disease protein, a human copper-transporting ATPase. J Biol Chem 276:36289–36294
Voskoboinik I et al (2003) Protein kinase-dependent phosphorylation of the Menkes copper P-type ATPase. Biochem Biophys Res Commun 303:337–342
Bartee MY, Ralle M, Lutsenko S (2009) The loop connecting metal-binding domains 3 and 4 of ATP7B is a target of a kinase-mediated phosphorylation. Biochemistry 48:5573–5581
Sazinsky MH et al (2007) Characterization and structure of a Zn2+ and [2Fe–2S]-containing copper chaperone from Archaeoglobus fulgidus. J Biol Chem 282:25950–25959
Rosen BP (2002) Transport and detoxification systems for transition metals, heavy metals and metalloids in eukaryotic and prokaryotic microbes. Comp Biochem Physiol A Mol Integr Physiol 133:689–693
Stokes DL, Auer M, Zhang P, Kuhlbrandt W (1999) Comparison of H+-ATPase and Ca2+-ATPase suggests that a large conformational change initiates P-type ion pump reaction cycles. Curr Biol 9:672–679
Yang Y, Mandal AK, Bredeston LM, Gonzalez-Flecha FL, Arguello JM (2007) Activation of Archaeoglobus fulgidus Cu(+)-ATPase CopA by cysteine. Biochim Biophys Acta 1768:495–501
Safaei R, Otani S, Larson BJ, Rasmussen ML, Howell SB (2008) Transport of cisplatin by the copper efflux transporter ATP7B. Mol Pharmacol 73:461–468
Odermatt A, Solioz M (1995) Two trans-acting metalloregulatory proteins controlling expression of the copper-ATPases of Enterococcus hirae. J Biol Chem 270:4349–4354
Wu CC, Rice WJ, Stokes DL (2008) Structure of a copper pump suggests a regulatory role for its metal-binding domain. Structure 16:976–985
Tsuda T, Toyoshima C (2009) Nucleotide recognition by CopA, a Cu+-transporting P-type ATPase. EMBO J 28:1782–1791
Mandal AK, Yang Y, Kertesz TM, Arguello JM (2004) Identification of the transmembrane metal binding site in Cu+-transporting PIB-type ATPases. J Biol Chem 279:54802–54807
Kuhlbrandt W (2004) Biology, structure and mechanism of P-type ATPases. Nat Rev Mol Cell Biol 5:282–295
Hatori Y, Majima E, Tsuda T, Toyoshima C (2007) Domain organization and movements in heavy metal ion pumps: papain digestion of CopA, a Cu+-transporting ATPase. J Biol Chem 282:25213–25221
Lubben M et al (2009) Structural model of the CopA copper ATPase of Enterococcus hirae based on chemical cross-linking. Biometals 22:363–375
Moller JV, Juul B, le Maire M (1996) Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim Biophys Acta 1286:1–51
Petris MJ et al (2002) Copper-regulated trafficking of the Menkes disease copper ATPase is associated with formation of a phosphorylated catalytic intermediate. J Biol Chem 277:46736–46742
Cater MA, La Fontaine S, Mercer JF (2007) Copper binding to the N-terminal metal-binding sites or the CPC motif is not essential for copper-induced trafficking of the human Wilson protein (ATP7B). Biochem J 401:143–153
Koonin EV, Tatusov RL (1994) Computer analysis of bacterial haloacid dehalogenases defines a large superfamily of hydrolases with diverse specificity. Application of an iterative approach to database search. J Mol Biol 244:125–132
Dmitriev O et al (2006) Solution structure of the N-domain of Wilson disease protein: distinct nucleotide-binding environment and effects of disease mutations. Proc Natl Acad Sci USA 103:5302–5307
Morgan CT, Tsivkovskii R, Kosinsky YA, Efremov RG, Lutsenko S (2004) The distinct functional properties of the nucleotide-binding domain of ATP7B, the human copper-transporting ATPase: analysis of the Wilson disease mutations E1064A, H1069Q, R1151H, and C1104F. J Biol Chem 279:36363–36371
Tsivkovskii R, Efremov RG, Lutsenko S (2003) The role of the invariant His-1069 in folding and function of the Wilson’s disease protein, the human copper-transporting ATPase ATP7B. J Biol Chem 278:13302–13308
Anthonisen AN, Clausen JD, Andersen JP (2006) Mutational analysis of the conserved TGES loop of sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem 281:31572–31582
Toyoshima C, Norimatsu Y, Iwasawa S, Tsuda T, Ogawa H (2007) How processing of aspartylphosphate is coupled to lumenal gating of the ion pathway in the calcium pump. Proc Natl Acad Sci USA 104:19831–19836
Sorensen TL et al (2004) Localization of a K+-binding site involved in dephosphorylation of the sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem 279:46355–46358
DiDonato M, Narindrasorasak S, Forbes JR, Cox DW, Sarkar B (1997) Expression, purification, and metal binding properties of the N-terminal domain from the wilson disease putative copper-transporting ATPase (ATP7B). J Biol Chem 272:33279–33282
Lutsenko S, Petrukhin K, Cooper MJ, Gilliam CT, Kaplan JH (1997) N-terminal domains of human copper-transporting adenosine triphosphatases (the Wilson’s and Menkes disease proteins) bind copper selectively in vivo and in vitro with stoichiometry of one copper per metal-binding repeat. J Biol Chem 272:18939–18944
Banci L, Bertini I, Cantini F, Rosenzweig AC, Yatsunyk LA (2008) Metal binding domains 3 and 4 of the Wilson disease protein: solution structure and interaction with the copper(I) chaperone HAH1. Biochemistry 47:7423–7429
Achila D et al (2006) Structure of human Wilson protein domains 5 and 6 and their interplay with domain 4 and the copper chaperone HAH1 in copper uptake. Proc Natl Acad Sci USA 103:5729–5734
Jones CE, Daly NL, Cobine PA, Craik DJ, Dameron CT (2003) Structure and metal binding studies of the second copper binding domain of the Menkes ATPase. J Struct Biol 143:209–218
Walker JM et al (2004) The N-terminal metal-binding site 2 of the Wilson’s Disease Protein plays a key role in the transfer of copper from Atox1. J Biol Chem 279:15376–15384
Banci L et al (2005) An atomic-level investigation of the disease-causing A629P mutant of the Menkes protein, ATP7A. J Mol Biol 352:409–417
DeSilva TM, Veglia G, Opella SJ (2005) Solution structures of the reduced and Cu(I) bound forms of the first metal binding sequence of ATP7A associated with Menkes disease. Proteins 61:1038–1049
Banci L, Bertini I, Del Conte R, D’Onofrio M, Rosato A (2004) Solution structure and backbone dynamics of the Cu(I) and apo forms of the second metal-binding domain of the Menkes protein ATP7A. Biochemistry 43:3396–3403
Gitschier J, Moffat B, Reilly D, Wood WI, Fairbrother WJ (1998) Solution structure of the fourth metal-binding domain from the Menkes copper-transporting ATPase. Nat Struct Biol 5:47–54
Tsay MJ, Fatemi N, Narindrasorasak S, Forbes JR, Sarkar B (2004) Identification of the “missing domain” of the rat copper-transporting ATPase, atp7b: insight into the structural and metal binding characteristics of its N-terminal copper-binding domain. Biochim Biophys Acta 1688:78–85
Cater MA, Forbes J, La Fontaine S, Cox D, Mercer JF (2004) Intracellular trafficking of the human Wilson protein: the role of the six N-terminal metal-binding sites. Biochem J 380:805–813
Mercer JF, Barnes N, Stevenson J, Strausak D, Llanos RM (2003) Copper-induced trafficking of the cU-ATPases: a key mechanism for copper homeostasis. Biometals 16:175–184
Strausak D et al (1999) The role of GMXCXXC metal binding sites in the copper-induced redistribution of the Menkes protein. J Biol Chem 274:11170–11177
Banci L et al (2009) An NMR study of the interaction of the N-terminal cytoplasmic tail of the Wilson disease protein with copper(I)-HAH1. J Biol Chem 284:9354–9360
Yatsunyk LA, Rosenzweig AC (2007) Cu(I) binding and transfer by the N terminus of the Wilson disease protein. J Biol Chem 282:8622–8631
Ralle M, Lutsenko S, Blackburn NJ (2004) Copper transfer to the N-terminal domain of the Wilson disease protein (ATP7B): X-ray absorption spectroscopy of reconstituted and chaperone-loaded metal binding domains and their interaction with exogenous ligands. J Inorg Biochem 98:765–774
Arnesano F et al (2002) Metallochaperones and metal-transporting ATPases: a comparative analysis of sequences and structures. Genome Res 12:255–271
O’Halloran TV, Culotta VC (2000) Metallochaperones, an intracellular shuttle service for metal ions. J Biol Chem 275:25057–25060
Banci L, Bertini I, Del Conte R, Markey J, Ruiz-Duenas FJ (2001) Copper trafficking: the solution structure of Bacillus subtilis CopZ. Biochemistry 40:15660–15668
Hamza I, Schaefer M, Klomp LW, Gitlin JD (1999) Interaction of the copper chaperone HAH1 with the Wilson disease protein is essential for copper homeostasis. Proc Natl Acad Sci USA 96:13363–13368
Huffman DL, O’Halloran TV (2001) Function, structure, and mechanism of intracellular copper trafficking proteins. Annu Rev Biochem 70:677–701
Puig S et al (2007) Higher plants possess two different types of ATX1-like copper chaperones. Biochem Biophys Res Commun 354:385–390
Andres-Colas N et al (2006) The Arabidopsis heavy metal P-type ATPase HMA5 interacts with metallochaperones and functions in copper detoxification of roots. Plant J 45:225–236
Wernimont AK, Huffman DL, Lamb AL, O’Halloran TV, Rosenzweig AC (2000) Structural basis for copper transfer by the metallochaperone for the Menkes/Wilson disease proteins. Nat Struct Biol 7:766–771
Ralle M, Lutsenko S, Blackburn NJ (2003) X-ray absorption spectroscopy of the copper chaperone HAH1 reveals a linear two-coordinate Cu(I) center capable of adduct formation with exogenous thiols and phosphines. J Biol Chem 278:23163–23170
Pufahl RA et al (1997) Metal ion chaperone function of the soluble Cu(I) receptor Atx1. Science 278:853–856
Banci L, Bertini I, Chasapis CT, Rosato A, Tenori L (2007) Interaction of the two soluble metal-binding domains of yeast Ccc2 with copper(I)-Atx1. Biochem Biophys Res Commun 364:645–649
Banci L, Bertini I, Ciofi-Baffoni S, Gonnelli L, Su XC (2003) Structural basis for the function of the N-terminal domain of the ATPase CopA from Bacillus subtilis. J Biol Chem 278:50506–50513
Larin D et al (1999) Characterization of the interaction between the Wilson and Menkes disease proteins and the cytoplasmic copper chaperone, HAH1p. J Biol Chem 274:28497–28504
Banci L et al (2007) The different intermolecular interactions of the soluble copper-binding domains of the Menkes protein, ATP7A. J Biol Chem 282:23140–23146
Bunce J, Achila D, Hetrick E, Lesley L, Huffman DL (2006) Copper transfer studies between the N-terminal copper binding domains one and four of human Wilson protein. Biochim Biophys Acta 1760:907–912
Rosenzweig AC et al (1999) Crystal structure of the Atx1 metallochaperone protein at 1.02 A resolution. Structure 7:605–617
Wernimont AK, Yatsunyk LA, Rosenzweig AC (2004) Binding of copper(I) by the Wilson disease protein and its copper chaperone. J Biol Chem 279:12269–12276
Banci L et al (2009) Copper(I)-mediated protein–protein interactions result from suboptimal interaction surfaces. Biochem J 422:37–42
Op’t Holt BT, Merz KM Jr (2007) Insights into Cu(I) exchange in HAH1 using quantum mechanical and molecular simulations. Biochemistry 46:8816–8826
Banci L et al (2006) The Atx1–Ccc2 complex is a metal-mediated protein–protein interaction. Nat Chem Biol 2:367–368
Singleton C et al (2008) Structure and Cu(I)-binding properties of the N-terminal soluble domains of Bacillus subtilis CopA. Biochem J 411:571–579
Gonzalez-Guerrero M, Eren E, Rawat S, Stemmler TL, Arguello JM (2008) Structure of the two transmembrane Cu+ transport sites of the Cu+-ATPases. J Biol Chem 283:29753–29759
Mandal AK, Arguello JM (2003) Functional roles of metal binding domains of the Archaeoglobus fulgidus Cu(+)-ATPase CopA. Biochemistry 42:11040–11047
Fan B, Grass G, Rensing C, Rosen BP (2001) Escherichia coli CopA N-terminal Cys(X)(2)Cys motifs are not required for copper resistance or transport. Biochem Biophys Res Commun 286:414–418
Strausak D et al (2003) Kinetic analysis of the interaction of the copper chaperone Atox1 with the metal binding sites of the Menkes protein. J Biol Chem 278:20821–20827
Lorinczi E et al (2008) Delivery of the Cu-transporting ATPase ATP7B to the plasma membrane in Xenopus oocytes. Biochim Biophys Acta 1778:896–906
Tsivkovskii R, MacArthur BC, Lutsenko S (2001) The Lys1010–Lys1325 fragment of the Wilson’s disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. J Biol Chem 276:2234–2242
Lutsenko S, Efremov RG, Tsivkovskii R, Walker JM (2002) Human copper-transporting ATPase ATP7B (the Wilson’s disease protein): biochemical properties and regulation. J Bioenerg Biomembr 34:351–362
Veldhuis NA et al (2009) Phosphorylation regulates copper-responsive trafficking of the Menkes copper transporting P-type ATPase. Int J Biochem Cell Biol (Epub ahead of print)
Valverde RH et al (2008) Cyclic AMP-dependent protein kinase controls energy interconversion during the catalytic cycle of the yeast copper-ATPase. FEBS Lett 582:891–895
Acknowledgments
This work was funded by National Institutes of Health grant R01 DK071865 to S.L. A.N.B. is a recipient of NRSA award F32DK077429.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article will be printed in the upcoming Journal of Biological Inorganic Chemistry special issue Cell Biology of Copper.
Rights and permissions
About this article
Cite this article
Barry, A.N., Shinde, U. & Lutsenko, S. Structural organization of human Cu-transporting ATPases: learning from building blocks. J Biol Inorg Chem 15, 47–59 (2010). https://doi.org/10.1007/s00775-009-0595-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-009-0595-4