
Abstract
The metal–thiolate connectivity of recombinant Cd7-MT10 metallothionein from the sea mussel Mytilus galloprovincialis has been investigated for the first time by means of multinuclear, multidimensional NMR spectroscopy. The internal backbone dynamics of the protein have been assessed by the analysis of 15N T 1 and T 2 relaxation times and steady state {1H}–15N heteronuclear NOEs. The 113Cd NMR spectrum of mussel MT10 shows unique features, with a remarkably wide dispersion (210 ppm) of 113Cd NMR signals. The complete assignment of cysteine Hα and Hβ proton resonances and the analysis of 2D 113Cd–113Cd COSY and 1H–113Cd HMQC type spectra allowed us to identify a four metal–thiolate cluster (α-domain) and a three metal–thiolate cluster (β-domain), located at the N-terminal and the C-terminal, respectively. With respect to vertebrate MTs, the mussel MT10 displays an inversion of the α and β domains inside the chain, similar to what observed in the echinoderm MT-A. Moreover, unlike the MTs characterized so far, the α-domain of mussel Cd7-MT10 is of the form M4S12 instead of M4S11, and has a novel topology. The β-domain has a metal–thiolate binding pattern similar to other vertebrate MTs, but it is conformationally more rigid. This feature is quite unusual for MTs, in which the β-domain displays a more disordered conformation than the α-domain. It is concluded that in mussel Cd7-MT10, the spacing of cysteine residues and the plasticity of the protein backbone (due to the high number of glycine residues) increase the adaptability of the protein backbone towards enfolding around the metal–thiolate clusters, resulting in minimal alterations of the ideal tetrahedral geometry around the metal centres.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metallothioneins (MTs) are a family of small proteins (6–7 kDa) with a remarkable affinity for metal ions with the d 10 electronic configuration, such as Zn(II), Cd(II) and Cu(I) [1, 2]. The metal binding capability is due to the high content of cysteine residues (typically accounting for about 30% of the amino acid composition), which form metal–thiolate coordinative bonds. MTs are ubiquitary proteins that have been found in a very wide range of organisms, including vertebrates, invertebrates, plants and bacteria [3]. The primary physiological roles of MTs are in homeostasis of essential trace elements as well as detoxification of toxic metals. Moreover, MTs seem to be involved in the protection of cells and tissues against various forms of oxidative injury caused by free radicals [4]. However, the detailed physiological functions of MTs are still matter of debate.
The earliest studies of MTs focused on mammalian isoforms, especially on the fully metallated Cd7- or Zn2Cd5-MT. The common features of mammalian MTs include: (1) a short polypeptidic chain of about 60 residues; (2) the presence of 20 cysteine residues arranged in motifs consisting of CC, CXC, and CXXC sequences; (3) the absence of aromatic residues. All mammalian Cd7-MTs are dumbbell-shaped monomers composed of two domains (each containing a metal–thiolate cluster) connected by a flexible linker consisting of a Lys–Lys segment. The N-terminal β-domain binds three divalent metal ions, whereas the C-terminal α-domain binds four divalent ions [5, 6]. The α-domain is said to be “adamantane-like” [2, 7], and is composed of 11 sulfur atoms belonging to the same number of cysteine residues (M4S11 cluster). In this cluster, there are five cysteines bridging two metal centres and six cysteines making a metal–thiolate bond with a single metal centre to complete the tetrahedral coordination geometry around each of the cadmium centres. The β-domain is termed “distorted chair-like,” and is composed of nine cysteine residues (M3S9 cluster) with three sulfur atoms bridging two metal centres and the remaining six connected to a single metal centre. In both kinds of clusters, the Cd(II) centres have a tetrahedral coordination geometry. MTs are known to lack substantial secondary structure in the absence of metals (the apothionein form), and to assume stable folding only upon metal binding [8]. In the folded form, the protein backbone wraps around each of the two metal–thiolate clusters without assuming any regular secondary structure motif. The experimental three-dimensional (3D) structures of MTs solved so far show that the two domains behave as independent units, in the sense that their relative orientation within the protein is not defined. The linker region connecting the two domains is found or assumed to be flexible [9].
Cd7-MTs from organisms other than mammals share many features in common with mammalian Cd7-MTs, but there are some peculiarities. Among vertebrates, fish MTs display the canonical αβ-domain organization found in mammals, with two asymmetric globular domains binding four and three divalent metal ions [10]. Among invertebrates, sea urchin MTs display the same type of metal–thiolate cluster organization, but the domains are inverted inside the chain (the β-domain is at the C-terminal and the α-domain at the N-terminal) [11]. In crustacean MTs, the two domains are symmetric and bind three divalent metal ions each [5, 12, 13]. Although not identical, these clusters are structurally similar to the β-domain of mammalian MTs. On the other hand, molluscan MTs display a large variety of structures consisting solely of α-domains, solely of β-domains, or of αββ-domains, in addition to the canonical αβ-domain structure [14]. Moreover, there are examples of shorter MTs, such as the MT from yeast that binds seven univalent d 10 metal ions in a single metal thiolate cluster [15].
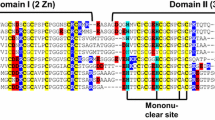
Whereas the amino acid sequences of many MTs from invertebrates have been identified, the complete 3D structures of only three MTs have been solved: the MT-1 isoforms from two crustaceans (the crab Callinectes sapidus [12] and the lobster Homarus americanus [16]) and the MT-A from the echinoderm Strongylocentrotus purpuratus [11]. No molluscan MT has had its 3D structure solved so far. Recently, the MT10 isoform from the sea mussel Mytilus galloprovincialis has been cloned and expressed as recombinant protein in Escherichia coli [17]. Two fully metallated forms (Zn7-MT10 and Cd7-MT10) have been produced and characterized in terms of metal binding stoichiometry, kinetics of metal release, scavenging activity, thermostability, and spectroscopic properties [18]. The metal binding stoichiometries of Cd(II) and Zn(II) indicate seven divalent metal ions bound per MT10 monomer, suggesting a similarity with mammalian and fish MTs. However, the functional and structural comparison of mussel MT10 with fish MT-A revealed a higher thermostability, a lower susceptibility to oxidative dimerization, and a greater reactivity of metal–thiolate clusters for the mussel MT10 protein. These features may correlate with the unusual primary sequence of mussel MT10 (Fig. 1a). Firstly, MT10 contains 21 cysteines, one more than the 20 cysteines typical of mammalian and fish MTs. Secondly, in line with other molluscan MTs, mussel MT10 contains a high number of glycine residues compared to MTs from other sources [19]. Finally, the primary sequence of MT10 shows no CC motifs. Since α-domains typically contain at least one CC sequence, whereas β-domains often lack of these motifs [11, 20], there is no obvious way to predict whether mussel MT10 is composed of canonical α-like and β-like domains, and to locate these domains within the primary structure. Some indication of a possible arrangement of MT10 into canonical domains has been obtained by aligning the primary structure of mussel MT10 with that of fish MT-A by means of the Needleman–Wunsch algorithm [17]. This procedure assigned a β-like domain at the N-terminal and an α-like domain at the C-terminal. However, this assignment presented some anomalies, namely in terms of the number of cysteines per protein domain (only eight for the β-domain, as many as 13 for the α-domain) and in the length of the linker (consisting of a single lysine residue). Therefore, an experimental assessment of the metal binding domains in mussel MT10 is still needed, and this constitutes the main focus of this work. NMR spectroscopy proved to be the technique of choice for the structural characterization of metal thiolate clusters in Cd7-MTs, given the molecular size of these proteins (almost ideal for this kind of study) and the possibility of taking advantage of the favourable NMR properties of the 113Cd nucleus [7]. We undertook the assignment of metal–thiolate connectivities by means of multinuclear, multidimensional protein NMR techniques, without using any a priori assumption about cluster location and topology. Moreover, 15N-spin relaxation properties were analyzed to assess internal protein dynamics and relate them with metal binding properties.

a Primary structure of Cd7-MT10, with the metal thiolate cluster connectivity determined by 1H–113Cd heterocorrelated NMR spectroscopy and 113Cd–113Cd COSY NMR spectroscopy. b Metal thiolate connectivities in the α-cluster. c Metal thiolate connectivity in the β-cluster. Residues marked with a minus sign are not part of the native form of MT10, and have been included for overexpression requirements. The residue marked with the letter X was changed to serine for overexpression requirements (it is a methionine in the native form). Residues marked with plus signs constitute the linker region between domains. The dotted line indicates an ambiguous assignment: it is not defined which of C9, C11 or C17 bridges between CdIII and CdV, even though all of them are connected to CdV
Experimental section
Materials
Chemicals, molecular weight markers and standard MT-I from rabbit were supplied by Sigma-Aldrich (Milan, Italy). Reagents for bacterial growth were purchased from Fluka (Milan, Italy). Expression vector pGEX 6P-1, E. coli strains and glutathione-Sepharose 4B matrix were from GE Healthcare Italy (Milan, Italy). Isotopically enriched 113Cd, 15N ammonium chloride and 13C-glucose were from Isotec–Sigma–Aldrich (Milan, Italy).
Cloning, expression and purification of MT-10
The full-length coding sequence of MT-10 gene from Mytilus galloprovincialis (GenBank accession no AY566248) was obtained as previously reported [17]. Through site-directed mutagenesis using primers designed ad hoc, we inserted a BamHI and an EcoRI site at the 5′-end of the encoding fragment and downstream from the stop codon, respectively. After the amplification reaction, the PCR fragments were digested with EcoRI and BamHI enzymes and subcloned in the pGEX-6P-1 vector, as previously described [17]. Recombinant MT-10 was expressed as fusion protein with a GST tail at the N′-terminus in the protease-deficient E. coli strain BL21. For large-scale expression, 12 mL of LB medium (10 g/L Tryptone, 5 g/L yeast extract, 5 g/L NaCl, 100 μg/mL ampicillin) were inoculated and grown overnight at 37 °C under vigorous shaking. One litre of pre-warmed 2XYT medium (16 g/L Tryptone, 10 g/L yeast extract, 5 g/L NaCl, 100 μg/mL ampicillin) was inoculated with 10 mL of the overnight culture and grown until it reached the mid-exponential growth phase. Following 5 h of induction with 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG, final concentration) at 30 °C, the cells were recovered by centrifugation at 4,000×g for 15 min and stored at −80 °C for further use.
To prepare selectively 13C- and 15N-labelled MT-10, bacteria were grown in minimal M9 medium supplemented with 0.1% 15N ammonium chloride and with 0.4% 13C glucose. For preparations of selectively 113Cd-labelled MT-10, 0.2 mM CdCl2 was added to the culture medium to produce 113Cd7-MT10.
The recombinant metallothionein was purified by affinity chromatography using glutathione-Sepharose 4B to selectively bind the GST tag of the fusion protein. The MT moiety was recovered by enzymatic cleavage, as previously described [17]. Recombinant MT-10 shows four additional amino acids (Gly-Pro-Leu-Gly) with respect to the wild-type protein, with the initial Met being substituted by a Ser.
Protein and metal quantification
The quality and purity of MT-10 were checked on 12.5% SDS-PAGE [37]. At each step of purification, the amount of total protein was determined by Bradford assay [38]. At the end of the purification procedure, the MT-10 concentration was determined spectrophotometrically via thiolate group quantification using Ellman’s reagent DTNB [5,5-dithiobis(2-nitrobenzoic acid)] with ε 412 = 13,600 M−1 cm−1 [17]. Rabbit MT-I was used as the standard for calibration. As an alternative, the metal-free protein was quantified via the absorption value at 220 nm under acidic conditions using ε 200 = 47,300 M−1 cm−1 [39]. The Cd2+ content of recombinant MT-10 was determined by atomic absorption spectroscopy using a polarized Spectra AA558 spectrometer (Varian Inc., Palo Alto, CA, USA) on proteins dissolved in 0.83 mM NaCl, 0.04 mM Tris–HCl pH 7, 0.1 mM HCl. A standard curve of CdCl2 was used for calibration.
NMR spectroscopy
All the NMR experiments were performed on a Bruker Avance 600 spectrometer operating at 14 T (corresponding to a proton Larmor frequency of 600.13 MHz). Double-resonance 1H–113Cd experiments as well as 1D 113Cd NMR spectra were carried out on a 113Cd7-MT10 sample enriched to 99% with 113Cd and dissolved in D2O (protein concentration 2.8 mM, Tris-d11 buffer 17 mM, pH 7.0, DTT-d10 16 mM, 298 K, D2O). 113Cd NMR chemical shifts were calibrated to external 0.1 M Cd(ClO4)2 in D2O. One-dimensional 113Cd NMR, 1D-saturation transfer and 2D 113Cd–113Cd COSY experiments were acquired with a Bruker BBO tunable probe in the temperature range 278–313 K (most often 298 K). The 2D 113Cd–113Cd COSY spectrum was obtained by a standard phase-insensitive COSY sequence with gradient coherence selection, with 2,048 acquired data points in F 2, 80 time increments in F 1, 1,024 scans, a 2 s recycle delay and a spectral window (both F 2 and F 1) of 400 ppm. Raw data were zero-filled to give a data matrix of 2,048 × 512 and multiplied by sine bell functions in both dimensions prior to FT and baseline correction. Proton-detected 113Cd–1H heteronuclear correlation spectra were collected either by standard gradient selected HMQC/HMBC pulse sequences (the latter including a delay of 60 ms for the evolution of long-range J-couplings) or by the modified HMQC sequence described by Frey et al. [24] to allow for relayed magnetization transfer between 113Cd and Cys Hα protons. To account for the large variability of 113Cd–1H coupling constants, these relayed HMQC experiments were performed with τ-delays of 15, 30, 40 and 60 ms. All proton-detected 113Cd–1H HMQC-type spectra were acquired with 2,048 data points in F 2 and 180 data points in F 1, 160 scans, a 2 s recycle delay, an F 2 (1H) spectral width of 6.0 ppm (carrier frequency at 3.2 ppm), and an F 1 (113Cd) spectral width of 300 ppm (carrier frequency at 571 ppm, i.e., exactly at the resonance of CdIII). Zero-filling to a data matrix of size 2,048 × 512 and multiplication by a π/4-shifted sine bell function in both F 2 and F 1 were performed before FT. 3D 1H/15N double-resonance and 3D 1H/15N/13C triple-resonance experiments [40] for backbone assignment were performed using a triple resonance inverse Bruker TXI probe with Z-axis PFG, at a temperature of 298 K. Either a 15N-labelled or a 15N/13C doubly labelled Cd7-MT10 sample at a 2 mM concentration in 95% H2O/D2O was used for these experiments (the samples also contained 17 mM Tris-d11 buffer pH 7.0, 10 mM DTT-d10). 1H–15N TOCSY HSQC, 1H–15N NOESY HSQC, and 15N-edited HNHA experiments were performed according to standard pulse sequences with water flip-back for water suppression, and with a data matrix of 1,024 × 48 × 220 complex points acquired in F 3(1H), F 2(15N) and F 1(1H), respectively. For 15N-edited TOCSY, mixing times of 50–100 ms were employed; for 15N-NOESY, mixing times of 50 and 120 ms were employed. For unambiguous backbone assignment, the following experiments were performed: HNCA, HN(CO)CA, CBCA(CO)NH, CBCANH and HBHA(CO)NH. All of these experiments were performed according to standard pulse sequences, with water flip-back for water suppression [41]. 15N longitudinal (T 1) and transverse (T 2) relaxation time constants and steady state {1H}–15N heteronuclear NOEs were measured on the globally 15N-labelled Cd7-MT10 sample described above at 298 K. Standard pulse sequences for T 1, T 2 and heteronuclear NOE were used [42], with 240 t 1 increments, 16 transients and 1,024 complex data points. In T 1 measurements, delays of 1,600, 20, 1,300, 50, 800, 100, 2,000, 200, 650, 150, 1,000, 370, 500 and 200 ms were used; in T 2 measurements, delays of 94.7, 11.8, 118.0, 35.5, 59.2, 142.8, 23.7, 47.4, 71.0, 165.8, 11.8 and 94.7 ms with a τ CPMG of 1.7 ms were used. The heteronuclear NOE experiment contained a 3.5 s proton presaturation period, whereas the reference no NOE experiment included a relaxation delay of 3.5 s. The multidimensional spectra were processed by Bruker XWINNMR 3.0 and resonance assignment via the XEASY software package [43]. Data processing of relaxation data was done by NMRpipe [44], and analysis of relaxation data by means of NMRView [45], by fitting T 1 and T 2 decays to a single-exponential function. Standard errors in relaxation time measurements were within 1% for T 1 and 3% for T 2. Steady-state NOE values were obtained by the ratio between peak volumes in NOE (1H saturation) and no NOE (no 1H saturation) experiments. Standard errors when measuring NOE were on average about 2%. Relaxation data analysis was performed according to the extended Lipari–Szabo formalism [26] by means of the MODELFREE 4.15 software package [46], by setting the 1H–15N average internuclear distance to 1.02 Å and the chemical shift anisotropy (Δσ) of amide 15N to −160 ppm (an axially symmetric chemical shift tensor was assumed).
Results
Overexpression and purification of recombinant Cd7-MT10
The recombinant protein produced in E. coli displays an additional tetrapeptide sequence (GPLG) at the N-terminal with respect to the wild-type protein [17]. Therefore, the recombinant MT10 is composed of 77 amino acids instead of the 73 that are present in the wild-type form. Throughout this manuscript, the numbering of amino acids in the primary structure of MT10 includes the additional tetrapeptide sequence. Thus, the first residue in the wild-type protein corresponds to residue number 5 in the recombinant protein. It is worth noting that such a residue (the first one in the native MT10 protein or the fifth according to the numbering scheme we adopted) was mutated from methionine to serine, again because of protein expression issues. The cadmium content in the recombinant MT-10 determined by atomic absorption spectrophotometry showed that the recombinant mussel MT-10 contains 7 equiv. of cadmium per mole of protein.
113Cd NMR spectroscopy
Figure 2 shows the proton-decoupled 113Cd NMR spectrum of Cd7-MT10 isotopically enriched to 99% with 113Cd (protein concentration 2.8 mM, Tris-d11 buffer 17 mM, pH 7.0, DTT-d10 16 mM, 298 K, D2O). This spectrum shows seven 113Cd NMR resonances falling within the 470–680 ppm chemical shift range, corresponding to the seven Cd(II) ions bound to the protein (resonances are numbered arbitrarily according to increasing chemical shifts). The signals from CdVI and CdVII show a multiplicity (doublet of doublet) due to coupling to two other cadmium ions (apparent values for both coupling constants are ~30 Hz). The signals from CdIV and CdV show a more complex multiplet structure, whereas the signals of CdI, CdII and especially that of CdIII appear somewhat broader. Interestingly, the 113Cd NMR resonances of MT10 are spread over a spectral window (210 ppm) that is much wider than that usually found for other mammalian and invertebrate Cd7-MTs (typically 90 ppm). As a matter of fact, all of the MTs characterized so far show 113Cd NMR resonance shifts that occur in the spectral window between 600 and 690 ppm [9, 10, 16, 21–24]. Saturation transfer 113Cd NMR experiments done by selectively saturating an individual 113Cd resonance and monitoring the change in intensities of the other 113Cd NMR resonances revealed no saturation transfer effects, indicating an absence of exchange on the second timescale. Moreover, 113Cd NMR spectra done within the temperature range 278–313 K showed only minor variations in the chemical shifts or linewidths of 113Cd NMR resonances, with a small broadening of the resonance of CdIII being the only detectable effect. All of these features strongly indicate that the cadmium binding domains in the protein are highly structured, with cadmium ions experiencing peculiar coordination environments, and that the structural features of the metal thiolate clusters may significantly differ from those currently known.

2D 113Cd–113Cd COSY NMR of Cd7-MT10 (protein concentration 2.8 mM, Tris-d11 buffer 17 mM, pH 7.0, DTT-d10 16 mM, 298 K, D2O). Cadmium atoms are labelled with Roman numerals according to increasing 113Cd chemical shift. The one-dimensional proton-decoupled 113Cd NMR spectrum is reported at the top. Expansions (4 ppm spectral window) of the individual cadmium resonances are given on the right
The homonuclear 2D 113Cd–113Cd COSY spectrum shown in Fig. 2 provided the first indication that the cadmium ions are grouped into two distinct metal thiolate clusters. On the one hand, this spectrum revealed cross-peaks connecting CdI with CdIII and CdV, and connecting CdIII with CdIV. On the other hand, CdII showed a correlation with CdVII, whereas CdVI showed no clear correlations above the noise. It must be noted that the 113Cd–113Cd COSY spectrum was characterized by a rather low signal-to-noise ratio (S/N), likely because of relaxation during the evolution time arising from the large CSA of 113Cd centres. Such an effect on the sensitivity of 113Cd–113Cd COSY spectroscopy has already been described [20]. By analogy with other MTs, the pattern of 113Cd–113Cd couplings prompted us to group CdI, CdIII, CdIV and CdV into a four-membered metal cluster and CdII, CdVI, CdVII into a three-membered metal cluster. In the latter cluster, CdVI should in principle give one correlation with CdVII and one correlation with CdII. The former should appear very close to the diagonal, and is expected to be completely masked by the CdVI and CdVII diagonal peaks. The CdVI–CdII correlation is likely missing because of low S/N. As explained below, the confirmation of the existence of a four-membered and a three-membered metal cluster as well as the details of the metal–thiolate connectivity within each of these clusters were provided by the analysis of the heterocorrelated 2D 1H–113Cd HMQC-type spectra.
Assignment of backbone HN, N, Cα, Cβ, Hα, Hβ resonances
A limitation of some previous NMR studies of Cd7-MTs was the inability to fully assign protein resonances because of a lack of properly labelled protein samples. This resulted in partial collection of the NMR data, in turn causing some loss of structural details or some ambiguity in the analysis of relaxation data. The protocol for the overexpression and purification of MT10 in minimal M9 medium containing isotopically enriched 15N-ammonium chloride and 13C-glucose as sources of carbon and nitrogen proved to be very effective, allowing for the isolation of a 15N–13C doubly labelled protein sample with high yields and a purity suitable for NMR spectroscopy. This enabled us to carry out triple-resonance 3D-NMR experiments [HNCA, HN(CO)CA, CBCA(CO)NH, CBCANH and HBHA(CO)NH] on Cd7-MT10, and to achieve the almost complete and unambiguous assignment of all of the HN, Cα, Cβ, Hα and Hβ resonances (Fig. 3 shows a fully assigned 1H–15N HSQC spectrum). Resonance assignment was further checked by double-resonance 3D-NMR experiments (1H–15N TOCSY HSQC, 1H–15N NOESY HSQC, 15N-edited HNHA) carried out on a 15N singly labelled Cd7-MT10 protein sample. The experimental conditions employed for the acquisition of both triple-resonance and double-resonance 3D-NMR spectra were 2.0 mM Cd7-MT10, pH 7.0, 17 mM Tris-d11 buffer, 10 mM DTT-d10, H2O:D2O 95%, T = 298 K. All of the cysteine residues showed resolved resonances for the diastereotopic Hβ pairs, except for C11, C19, C36 (a single resonance for both Hβ protons was detected). The assignment of the Hα and Hβ atoms of the cysteines thus obtained was used as the basis for discerning the connectivity between cysteines and cadmium ions through heterocorrelated inverse-detected 1H–113Cd 2D-NMR experiments.

2D 1H–15N HSQC NMR spectrum of 15N-labelled Cd7-MT10 (protein concentration 2.0 mM, 17 mM Tris-d11 buffer, pH 7.0, 10 mM DTT-d10, 298 K, H2O:D2O 95%). The assignment of backbone amide 1H–15N correlations is shown. Side chain correlations (folded) are denoted by sc
Assignment of heterocorrelated 2D 1H–113Cd NMR spectra
A series of standard heterocorrelated inverse-detected 1H–113Cd HMQC and HMBC spectra (with several delays of the low-pass filter for the detection of small 1H–113Cd couplings) have been acquired on a 113Cd7-MT10-enriched sample. Although the HMBC-type spectra gave the highest signal intensities, it was not possible to fully derive the connectivity between cadmium centres and cysteine residues because of severe overlap in the region containing the Cys Hβ resonances. In these HMBC spectra, a few correlations between Cys Hα and 113Cd could be detected (namely those between C23–CdIV, C76–CdVI, C58–CdVII, and C41–CdIII). Such additional Hα–113Cd correlations are very useful for elucidating the connectivities in the metal thiolate clusters, as the resonances of Cys Hα protons are better resolved than those of Cys Hβ protons [25]. To achieve the maximum number of Hα–113Cd correlations, the relayed version of the 1H–113Cd HMQC pulse sequence described by Frey et al. [24] was implemented with gradient selection. A series of 2D 1H–113Cd relayed HMQC spectra were acquired with polarization transfer τ delays of 15, 30, 40 and 60 ms (Fig. 4 reports a representative spectrum, obtained with τ = 30 ms), providing a wealth of Cys Hα–113Cd correlations that allowed us to define the pattern of 113Cd–cysteine connectivities without any a priori assumption about the topology of the metal clusters and their location within the polypeptide backbone. Consistent with the conclusions obtained by 2D 113Cd–113Cd COSY, it was found that two metal thiolate clusters are present in Cd7-MT10, one of the form Cd3S9 and one of the form Cd4S12. The former cluster is formed by the cysteine residues in the protein segment 50–77, and is thus located in the C-terminal portion of the protein. C58, C62 and C76 bridge between two cadmium centres, whereas the remaining six cysteines in this protein segment make one metal–thiolate bond with a single metal centre (Fig. 1c). The number of 1H–113Cd correlations found in heterocorrelated NMR spectra was large enough to provide an unambiguous assignment. The pattern of metal–thiolate bonds of the M3S9 cluster is essentially analogous to that forming the β-domain in a variety of typical MTs from either vertebrate or invertebrate organisms. The Cd4S12 cluster is located at the N-terminal (protein segment 5–46), with four cysteine thiolates bridging between two cadmium ions, and the remaining eight cysteine thiolates coordinating a single metal centre (Fig. 1b). Three bridging cysteines have been unambiguously identified (C30, C43 and C46), whereas the presence of a fourth bridging cysteine is inferential. This conclusion has been drawn by considering that each of CdI, CdIV and CdV showed unambiguous correlations with four cysteines, whereas CdIII showed clear correlations only with three cysteines (C41, C43 and C46). Amongst the correlations involving CdIII, those with C41(Hα) and C41(Hβ) were clearly seen in the relayed HMQC-type spectrum acquired with a τ-delay of 30 ms (shown in Fig. 4), whereas that with C43(Hα) was best observed with a τ-delay of 40 ms, and those with C46(Hβ) and C43(Hβ) were best detected with a τ-delay of 15 ms (not shown). It should be noted that the resonance of CdIII is characterized by quite a large linewidth (Fig. 2). To complete the coordination around CdIII, it was considered that one of the cysteines coordinating CdV (except C30, which is already engaged in bridging between CdV and CdI) should form an additional metal–thiolate bond with CdIII. Cysteine residues bound to CdI or CdIV cannot form a metal–thiolate bond with CdIII because this would result in very constrained and unlikely structures (under these circumstances, one pair of cadmium centres would present a double metal-to-metal cysteine thiolate bridge). Hence, one cysteine amongst C9, C11 or C17 is inferred to bridge between CdIII and CdV, as sketched in Fig. 1a, b. Following the convention adopted for mammalian MTs, we will refer to the protein domain containing the four-metal centre in Cd7-MT10 as to the α-domain, even though it has unique features when compared with the α-domain of typical MTs. In fact, one of the two rings in the adamantane-like structure is broken, and the completion of the tetrahedral coordination around the CdI and CdIV centres is made possible by the “extra” cysteine (i.e. the 21st cysteine, which exceeds the ideal number of 20 that are typically needed to make one canonical three-metal and one canonical four-metal cluster).

1H–113Cd HMQC with relayed Hα–Hβ magnetization transfer (τ delay = 30 ms) of 113Cd-enriched Cd7-MT10 (protein concentration 2.8 mM, Tris-d11 buffer 17 mM, pH 7.0, DTT-d10 16 mM, 298 K, D2O). Visualization as strips centered at each of the 113Cd resonances is provided, along with assignments of the most relevant 1H–113Cd correlations. 1Hα–113Cd correlations are given in bold; bridging cysteines are given in italics
Backbone dynamics via 15N spin relaxation
15N longitudinal (T 1) and transverse (T 2) relaxation time constants together with steady-state {1H}–15N heteronuclear NOEs were measured on a 15N-labelled Cd7-MT10 at 298 K for all residues, in both the β- and the α-domains. These values are plotted against residue number in Fig. 5. The N-terminal octapeptide shows large negative NOEs along with T 2 relaxation times that are much longer than the protein average, indicating that it is essentially unfolded and very flexible. This is expected, since a large part of this fragment is not part of the native molecule and does not fold around the metal centres. Therefore, this octapeptide is excluded from further discussion. The relaxation parameters averaged over the whole protein (segment C9–K77) show values of 151.1 ± 29.8 ms for T 2, 512.7 ± 60.0 ms for T 1 and 0.64 ± 0.13 for the heteronuclear NOE (see Table 1). These values do not change appreciably when averaged over the residues defining the α-domain (C9–C64) or the β-domain (C50–K77), indicating that both protein domains are characterized by similar backbone dynamics. Despite the overall homogeneity of internal motions within the protein backbone, it is clear from Fig. 5 that there are short regions characterized by a reduction in the magnitude of the heteronuclear NOE and by the simultaneous increase of T 2 and T 1 relaxation times. Two of these regions are located within the α-domain (T21–C23, S37–C41), whereas a third region is located within the β-domain (S53–S55). The reduction of NOEs coupled with the increase in T 2 can be taken as evidence of the internal dynamics of the backbone at the subnanosecond timescale. Remarkably, the interdomain linker region (K47–V49) shows dynamics in line with the protein average. Therefore, the backbone dynamics within the two domains and the linker region appear to be rather homogeneous, with the exception of short segments endowed with local disorder, which is dynamic in nature.

Plot of backbone 15N amide T 2 (a), T 1 (b), heteronuclear NOEs (c) and square generalized order parameter S 2 (d) as a function of residue number in mussel Cd7-MT10 (data obtained at 298 K). The vertical lines indicate the different domains of the protein
To get a more quantitative picture, the 15N relaxation data were analyzed according to the model-free formalism of Lipari and Szabo [26], which describes the motions that cause relaxation in terms of a spectral density function that basically contains three parameters: an isotropic rotational correlation time (τ m), an effective correlation time describing the rapid internal motions (τ e), and a generalized order parameter measuring the amplitude of internal motions (S 2). Under the assumptions of (1) a low degree of rotational anisotropy and (2) limited (S 2 approaching unity) and fast (t e ≪ 100 ps) internal motions, the spectral density function can be simplified such that T 1/T 2 ratios can be fitted on a residue-by-residue basis and then averaged to extract the overall rotational correlation time τ m [27, 28]. Residues that might be involved in large-amplitude, fast motions, identified by negative or low NOE values (NOE < 0.65, residues T21–C23, S37–C41, S53–S55), or those involved in chemical exchange processes, identified by T 1/T 2 ratios that are greater than one standard deviation from the mean (R69), were excluded from such analysis. As summarized in Table 1, the global τ m obtained by averaging over the protein segment 9–77 was 4.9 ns; this value is very close to those obtained by considering T 1/T 2 ratios of residues belonging only to the α-domain (5.0 ns) or only to the β-domain (4.7 ns). Further analysis of the relaxation data was performed according to the local site treatment [29, 30], according to which each spin is treated as behaving like an isotropic tumbler characterized by its own rotational correlation time τ m,eff. The parameters S 2, τ m,eff and τ e are each fitted as local dynamics parameters, and the isotropic, global τ m can then be obtained from an average of local correlation times. This approach offers the advantage of directly including the NOE data in calculations, thus improving the accuracy of the results. In addition, the fitting of model free parameters should not be significantly affected by moderate anisotropic tumbling, which could be the case for MTs. Most of the residues showed a good fit to this model, but residues C9, K35, C36, V49, K51 and R69 showed somewhat larger fitting errors. These residues are characterized by higher T 1/T 2 ratios, indicating that their relaxations might have a significantly contribution from chemical exchange processes. These residues, together with those showing local disorder (S 2 < 0.75), were excluded from the calculation of global correlation times. The global correlation times obtained by averaging τ m,eff over the whole protein, the α-domain and the β-domain were the same as those obtained by the global approach (Table 1). The plot of S 2 against the protein sequence confirms the dynamic picture of the protein backbone given above in qualitative terms. The majority of protein residues show S 2 > 0.8, whereas some peaks of low S 2 (below 0.6) are found in segments T21–C23, G38–C41, and around residue G54. This confirms that the α- and β-domains are characterized by comparable backbone rigidity, with flexible regions being confined to very short loop segments. Noticeably, the linker region between the two domains is as rigid as the structured residues that make up the metal thiolate clusters, suggesting a fixed relative orientation of the two protein domains.
The backbone dynamics of Cd7-MT10 show quite new features in comparison to other vertebrate and invertebrate Cd7-MTs composed of one α-domain and one β-domain. In Cd7-MT10, the α- and β-domains show similar internal dynamics overall, whereas a higher mobility (disorder) of the backbone in the β-domain with respect to the α-domain is typical of MTs [9], including mammalian MT-1/MT-2 [2], MT-3 [22, 31], fish MT_nc [10, 32], and sea urchin MTA [11]. Actually, the high flexibility of the backbone in the β-domains of these MTs causes the collapse of amide signals into regions of severe spectral overlap, preventing the full assignment of backbone resonances.
Discussion
The full analysis of 1H–113Cd NMR spectra of mussel Cd7-MT10 allowed for the unambiguous localization of the α-domain at the N-terminal (segment 9–46) and the β-domain at the C-terminal (segment 50–77), the two domains being separated by a short KVV linker. This arrangement is inverted with respect to canonical vertebrate MTs and resembles that of echinoderm MT A [11], in which the inverted sequential order of the protein domains has been related to the different gene structures (inversion of the order of the second and third exons in mammalian and echinoid [33]). In this regard, we must rectify previous conclusions about the sequence of the cadmium-binding domains within the protein, based upon sequence alignment and comparative analysis of fish MT-A and mussel MT10 [17, 18].
The unusual features of the primary structure of mussel Cd7-MT10 (high content of glycine residues, presence of 21 cysteine residues instead of 20, and absence of CC motifs) are paralleled by unique spectroscopic features. Mussel Cd7-MT10 shows unusually dispersed 113Cd NMR signals, with some resonances being shifted significantly upfield. Namely, the resonances of CdI and CdIII (belonging to the α-domain) fall at 463.5 and 571.8 ppm, respectively, whereas that of CdII (belonging to the β-domain) falls at 521.1 ppm. It is worth noting that 113Cd NMR resonances in Cd7-MTs usually fall within the 600–690 ppm spectral range. This suggests that the tridimensional structure and/or the coordination environment around the cadmium centres might be significantly different from those known so far. The most evident peculiarity of mussel Cd7-MT10 in comparison with other MTs is the pattern of metal thiolate bonds in the α-domain. In mussel Cd7-MT10, the α-cluster is of the form M4S12 instead of M4S11, with the extra cysteine making an additional coordination bond with a single cadmium centre. As a consequence, a bridging cysteine is transformed into a nonbridging one to maintain the tetrahedral coordination around each of the cadmium centres. This leads to the opening of one of the two fused six-membered metal thiolate rings that make the adamantane-like cluster (Fig. 1b). The pattern of metal thiolate bonds in the β-domain is analogous to that of classical MTs, but the unusual upfield shift of the CdII resonance still indicates some peculiarity. It has been pointed out that 113Cd NMR chemical shifts are exceedingly sensitive to very subtle changes in the coordination geometry around the metal centre in MTs [7] and other metalloproteins containing the Cd(S–Cys)4 metal thiolate group [34]. For tetrathiolate metalloproteins, a relationship has been found between the geometry at the metal centres and the observed 113Cd NMR chemical shifts. Namely, the chemical shift moves downfield as the distortion of the tetrahedral geometry at the 113Cd(S–Cys)4 metal centre increases, due to an increase of the paramagnetic deshielding contribution to the chemical shift tensor [34]. Following this line, the upfield shift of CdI–CdIII (belonging to the α-cluster) and CdII (belonging to the β-cluster) indicates that these metal centres are endowed with minimal distortions of the tetrahedral coordination geometry. This can be related to the presence of a high number of glycine residues in the protein sequence (as many as seven in the α-domain and four in the β-domain), which is expected to result into an increased conformational freedom and plasticity of the protein backbone. The analysis of 15N-spin relaxation parameters clearly shows that the internal backbone dynamics of the protein are essentially homogeneous over the whole protein, with increased degrees of internal motions being confined to very short segments. Hence, the structural function of glycine does not appear to enhance the dynamic disorder in the structure, but rather to increase the adaptability of the protein backbone towards the enfolding around the metal thiolate clusters. The spacing of cysteine residues and the conformational flexibility of the protein backbone of MT10 appears to be finely tuned to accommodating metal thiolate clusters with minimal alterations of the ideal tetrahedral geometry. These clusters fit well into the protein fold and are endowed with quite a rigid structure, as evidenced by the absence of inter- or intra-domain metal exchange (saturation transfer experiments).
There are several indications of the possibility of a unique domain arrangement in mussel Cd7-MT10, in which the two lobes interact (correlated motions) or even adopt a preferential relative orientation. Namely: (1) both the α- and the β-protein domains are highly structured (high dispersion of 1H and 113Cd NMR signals); (2) the two protein domains show comparable internal dynamics (homogeneous distribution of relaxation times and S 2 over the whole protein length); (3) the inter-domain linker does not show conformational flexibility. The analysis of correlation times for overall reorientation might provide further insights into this point. Despite the different sizes of the α- and β-domains (approximately 38 and 23 residues, respectively), both domains show τ m values close to 5 ns, with the percentage difference between correlation times being as small as 6%. As a reference for fully independent motions of the protein domains, one can calculate τ m values for the isolated domains according to an empirical relationship linking the molecular size of a protein with τ m (isotropic tumbling is assumed) [35]. τ m values of 3.0 and 2.3 ns for the α-domain and β-domain, respectively, can thus be obtained, corresponding to a percentage difference of 23%. As a reference for the τ m expected for a globular protein having the size of whole Cd7-MT10, a value of 5.4 ns can be obtained on the basis of the same empirical relationship. On these grounds, it would be concluded that the protein domains are not dynamically independent. However, it has been shown that in proteins that are known to contain two dynamically independent domains (fully uncorrelated motions), the presence of one domain still affects the correlation time of the other domain, and the actual difference in the correlation times between the two domains is much smaller than expected based on the difference in size [31, 36]. It should finally be noted that, in human MT-3, residues within structured regions in the β-domain showed a global τ m value similar to that for the α-domain, even though very different internal dynamics for the two domains and an absence of correlated domain motions were found [22, 31]. It is therefore difficult to assess whether the observed difference of 6% between the correlation times of the two domains of Cd7-MT10 is truly indicative of a motionally restricted arrangement of the protein lobes, and conclusive evidence is still needed.
References
Kagi JHR (1991) Methods Enzymol 205:613–626
Romero-Isart N, Vasak M (2002) J Inorg Biochem 88:388–396
Kagi JHR (1993) In: Suzuki K, Imura N, Kimura M (eds) Metallothionein III. Birkhauser-Verlag, Basel, pp 29–55
Bremner I (1991) Methods Enzymol 205:25–35
Otvos JD, Olafson RW, Armitage IM (1982) J Biol Chem 257:2427–2431
Braun W, Vasak M, Robbins AH, Stout CD, Wagner G, Kagi JHR, Wuthrich K (1992) Proc Natl Acad Sci USA 89:10124–10128
Vasak M (1998) Biodegradation 9:501–512
Willner H, Vasak M, Kagi JHR (1987) Biochemistry 26:6287–6292
Zangger K, Armitage IM (2002) J Inorg Biochem 88:135–143
Capasso C, Carginale V, Crescenzi O, Di Maro D, Parisi E, Spadaccini R, Temussi PA (2003) Structure 11:435–443
Riek R, Precheur B, Wang Y, Mackay EA, Wider G, Gunthert P, Liu A, Kagi JHR, Wuthrich K (1999) J Mol Biol 291:417–428
Narula SS, Brouwer M, Hua Y, Armitage IM (1995) Biochemistry 34:620–631
Overnell J, Good M, Vasak M (1988) Eur J Biochem 172:171–177
Jenny MJ, Ringwood AH, Schey K, Warr GW, Chapman RW (2004) Eur J Biochem 271:1702–1712
Peterson CW, Narula SS, Armitage IM (1996) FEBS Lett 379:85–93
Munoz A, Forsterling FH, Shaw CFIII, Petering DH (2002) J Biol Inorg Chem 7:713–724
Vergani L, Grattarola M, Borghi C, Dondero F, Viarengo A (2005) FEBS J 272:6014–6023
Vergani L, Grattarola M, Grasselli E, Dondero F, Viarengo A (2007) Arch Biochem Biophys 465:247–253
Barsyte D, White KN, Lovejoy DA (1999) Comp Biochem Physiol C Toxicol Pharmacol 122:287–296
Wang Y, Mackay EA, Zerbe O, Hess D, Hunziker PE, Vasak M, Kagi JHR (1995) Biochemistry 34:7460–7467
Messerle BA, Schaffer A, Vasak M, Kagi JHR, Wuthrich K (1990) J Mol Biol 214:765–779
Wang H, Zhang Q, Cai B, Li H, Sze K-H, Huang Z-X, Wu H-M, Sun H (2006) FEBS Lett 580:795–800
Shultze P, Worgotter E, Braun W, Wagner G, Vasak M, Kagi JHR, Wuthrich K (1988) J Mol Biol 203:251–268
Frey MH, Wagner G, Vasak M, Sorensen OW, Neuhaus D, Worgotter E, Kagi JHR, Ernst RR, Wuthrich K (1985) J Am Chem Soc 107:6847–6851
Neuhaus D (2003) Magn Res Chem 41:S70–S79
Lipari G, Szabo A (1982) J Am Chem Soc 104:4546–4559
Kay LE, Torchia DA, Bax A (1989) Biochemistry 28:8972–8979
Clore GM, Driscoll PC, Wingfield PT, Gronenborn AM (1990) Biochemistry 29:7387–7401
Lee AL, Wand AJ (1999) J Biomol NMR 13:101–112
Palmer AGIII (2001) Annu Rev Biophys Biomol Struct 30:129–155
Oz G, Zangger K, Armitage IM (2001) Biochemistry 40:11433–11441
D’Auria S, Carginale V, Scudiero R, Crescenzi O, Di Maro D, Temussi PA, Parisi E, Capasso C (2001) Biochem J 354:291–299
Harlow P, Watkins E, Thornton RD, Nemer M (1989) Mol Cell Biol 9:5445–5455
Goodfellow BJ, Joao Lima M, Ascenso C, Kennedy M, Sikkink R, Rusnak F, Moura I, Moura JJG (1998) Inorg Chim Acta 273:279–287
Daragan VA, Mayo KH (1997) Prog Nucl Magn Res Spectrosc 31:63–105
Campos-Olivas R, Newman JL, Summers MF (2000) J Mol Biol 296:633–649
Laemmli UK (1970) Nature 227:680–685
Bradford MM (1976) Anal Biochem 72:142–146
Bühler R, Kägi JHR (1978) Exp Suppl 34:211–220
Cavanagh J, Fairbrother WJ, Palmer AGIII, Skelton NJ (1996) Protein NMR spectroscopy. Academic, New York
Sattler M, Schleucher J, Griesinger C (1999) Prog NMR Spectrosc 34:93–158
Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE (1994) Biochemistry 33:5984–6003
Bartels C, Xia T-H, Billeter M, Güntert P, Wüthrich K (1995) J Biomol NMR 6:1–10
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax S (1995) J Biomol NMR 6:277–293
Johnson BA, Blevins RA (1994) J Biomol NMR 4:603–614
Palmer AGIII, Rance M, Wright PE (1991) J Am Chem Soc 113:4371–4380
Acknowledgments
The authors gratefully acknowledge Dr. G. Musco and Dr. L. Mollica (DIBIT, Milan) for helpful discussions, and Professor M. Piccioli (CERM, University of Florence) for allocating instrumental time under the Large Scale Facility programme. We would like to extend our gratitude to Myriam Grattarola and Mara Carloni for their experimental contributions, and to Professor Capannelli for allocating instrument time for the polarized Spectra AA558.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Digilio, G., Bracco, C., Vergani, L. et al. The cadmium binding domains in the metallothionein isoform Cd7-MT10 from Mytilus galloprovincialis revealed by NMR spectroscopy. J Biol Inorg Chem 14, 167–178 (2009). https://doi.org/10.1007/s00775-008-0435-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-008-0435-y