
Abstract
A binuclear copper complex, [Cu2(BPMP)(OAc)2][ClO4]·H2O, has been prepared using the binucleating ligand 2,6-bis[bis(pyridin-2-ylmethylamino)methyl]-4-methylphenol (H-BPMP). The X-ray crystal structure reveals the copper centers to have a five-coordinate square pyramidal geometry, with the acetate ligands bound terminally. The bridging phenolate occupies the apical position of the square-based pyramids and magnetic susceptibility, electron paramagnetic resonance (EPR) and variable-temperature variable-field magnetic circular dichroism (MCD) measurements indicate that the two centers are very weakly antiferromagnetically coupled (J = −0.6 cm−1). Simulation of the dipole–dipole-coupled EPR spectrum showed that in solution the Cu–O–Cu angle was increased from 126° to 160° and that the internuclear distance was larger than that observed crystallographically. The high-resolution spectroscopic information obtained has been correlated with a detailed ligand-field analysis to gain insight into the electronic structure of the complex. Symmetry arguments have been used to demonstrate that the sign of the MCD is characteristic of the tetragonally elongated environment. The complex also displays catecholase activity (k cat = 15 ± 1.5 min−1, K M = 6.4 ± 1.8 mM), which is compared with other dicopper catechol oxidase models.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Binuclear metalloenzymes are prevalent in nature, performing a range of functions on various substrates [1]. Dimeric copper sites play an important role in the activation of biological oxygen [2–4], and the study of structural and functional aspects of copper metalloenzymes via model systems is a subject of intense research [5–10]. A member of the family of dicopper proteins is catechol oxidase, which features a type 3 copper center with two proximate copper ions coordinated primarily by histidine donors [11–13]. This enzyme catalyzes the two-electron oxidation of o-diphenols to the corresponding quinones. The X-ray crystal structure of the oxidized catechol oxidase from sweet potato reveals a nitrogen-rich coordination environment, with three histidine donors to each copper [14]. The geometry about both copper centers is trigonal pyramidal. In the oxidized Cu(II)–Cu(II) form the metal–metal distance is 2.9 Å, while in the reduced form this distance increases to 4.4 Å. The mechanism proposed for the catechol oxidases involves the oxidation of two molecules of substrate per molecule of oxygen [13, 14]. It is proposed that one molecule of catechol binds, and is oxidized along with the two-electron reduction of the dicopper center (Fig. 1). Subsequently, a second molecule of substrate binds, along with one molecule of oxygen (bound as a peroxide in a μ–η2:η2 mode with a metal–metal distance of 3.8 Å determined by extended X-ray absorption fine structure (XAFS) [12]), followed by the reoxidation of the copper center. The catechol is then oxidized and released, with regeneration of the active site. The precise binding mode of the catechol is debated, with Eicken et al. [12] proposing monodentate coordination, while Solomon et al. [2] hypothesize a bidentate binding mode of the catechol.

Proposed catalytic cycle of catechol oxidase. (Redrawn from [13])
Design and study of model complexes for catechol oxidase and other type 3 copper centers has been an area of much interest [6–10] and various nitrogen-containing binucleating ligands have been used to generate dicopper complexes [6, 15–18]. 2,6-Bis[bis(pyridin-2-ylmethylamino)methyl]-4-methylphenol (H-BPMP), employed previously to model catechol oxidase [19, 20], provides a nitrogen-rich coordination set similar to that of the enzyme (three nitrogen donors) and has previously furnished both trigonal bipyramidal and square pyramidal geometries. A hydroxo-bridged dicopper complex with BPMP− has been shown to display catecholase activity [19]. Various complexes with other terminal ligating species have been characterized [19, 21, 22]. Several structure–activity relationships have been identified from amongst the previously reported catechol oxidase models. Specifically, a shorter copper–copper distance and an accessible redox potential appear to enhance reactivity, although these relationships are not straightforward [5, 9]. Typically, however, the spectroscopy of these dicopper model complexes has not been investigated in detail.
Many other binuclear enzymes have been modeled with bridging acetate moieties, which are displaced under kinetic conditions [23–27]. Reported here is the structure and catecholase activity of a dicopper complex of BPMP− with terminal, rather than bridging, acetate ligands, and the spectroscopic characterization thereof via magnetic circular dichroism (MCD), electron paramagnetic resonance (EPR) and magnetic susceptibility.
Materials and methods
All reagents were of analytical grade and were purchased from Sigma-Aldrich unless otherwise stated. H-BPMP was prepared according to literature methods [19].
Caution! Perchlorate salts of metal complexes are potentially explosive and therefore should be prepared in small quantities.
Preparation of [Cu2(BPMP)(OAc)2][ClO4]·H2O (1)
A solution of H-BPMP (240 mg, 0.45 mmol), copper acetate hydrate (165 mg, 0.9 mmol) and sodium perchlorate hydrate (63 mg, 0.45 mmol) in methanol (30 ml) was stirred at 313 K for 30 min. The dark green mixture was then allowed to stand at room temperature. A blue/green microcrystalline precipitate formed (270 mg, 67%). Slow evaporation of an acetonitrile solution yielded diffraction-quality crystals of [Cu2(BPMP)(OAc)2][ClO4]·H2O (1). Elemental analysis: calculated for C37H41N6O10ClCu2: C, 49.80; H, 4.63; N, 9.42. Found C, 49.70; H, 4.42; N, 9.15%.
IR spectroscopy was performed with a PerkinElmer Spectrum 2000 Fourier transform IR spectrometer with a Smiths DuraSamplIR II attenuated total refelection diamond window. Absorption spectra were measured with a Varian Cary50 Bio UV/vis spectrophotometer using 10-mm quartz cuvettes. Magnetic susceptibility studies in solution were undertaken with a Bruker AMX500 instrument at 298 K for a sample dissolved in acetonitrile. Appropriate diamagnetic corrections were used [28].
Single-crystal X-ray structure determination
Cell constants were determined by a least-squares fit to the setting parameters of 25 independent reflections measured with an Enraf–Nonius CAD4 four-circle diffractometer employing graphite monochromated Mo Kα radiation (0.71073 Å) and operating in the ω − 2θ scan mode. Data reduction was performed with the WINGX [29] package. Crystallographic data are reported in Table 1. The structure was solved using SIR92 [30] and refined using full-matrix least-squares analysis against F 2 with SHELXL-97 [31]. The hydrogen atom of the water solvate was located from the difference map. All non-hydrogen atoms were refined with anisotropic displacement parameters. The perchlorate counterion was disordered. Drawings of molecules were produced with ORTEP3 [32].
Crystallographic data (without structure factors) for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-65358. Copies of the data can be obtained free of charge from the CCDC (12 Union Road, Cambridge CB2 1EZ, UK; Tel: +44-1223-336408; Fax: +44-1223-336003; e-mail: deposit@ccdc.cam.ac.uk; Web site: http://www.ccdc.cam.ac.uk).
EPR spectroscopy
Continuous-wave X-band EPR spectra of 0.1 mM solutions of 1 in methanol were recorded at 120 K, 2-mW power, using a Bruker Biospin Elexsys E580 EPR spectrometer fitted with a super high Q cavity. Magnetic field and microwave frequency calibration were achieved with a Bruker ER 036 M Teslameter and a Bruker microwave frequency counter, respectively. Temperatures were controlled using a flow-through cryostat in conjunction with a Eurotherm (B-VT-2000) variable-temperature controller (120–140 K). Spectrometer tuning, signal averaging and subsequent spectral comparisons were performed with Bruker’s Xepr (version 2.3) software. Computer simulation of the EPR spectrum was performed using Molecular Sophe (version 2.0.97) running on a personal computer with the Mandriva operating system (2007.0). Figures were generated using gnuplot (http://www.gnuplot.info).
Magnetic susceptibility
Magnetic susceptibility measurements were made at the School of Chemistry, Monash University, Australia, using a Quantum Design MPMS SQUID magnetometer with an applied field of 1 T as a function of temperature (ranging from 2 to 300 K). The crystalline samples were enclosed in a calibrated gelatin capsule positioned in the center of a drinking straw fixed to the end of the sample rod. Effective magnetic moments, per mole, were calculated using the relationship μ eff = 2.828(χ m T)1/2, where χ m is the susceptibility per mole of complex. Fitting of the experimental magnetic susceptibility data was performed using Octave (http://www.gnu.org/software/octave/) and the results were plotted with gnuplot.
Magnetic circular dichroism
MCD spectra of a 4.8 mM solution of 1 in an ethanol/methanol (50:50) glass in a 1-mm path-length quartz cell were measured at ±5 T with an Oxford Instruments Spectromag instrument using a Hamamatsu R669 photomultiplier tube detector, with a Lastek-designed MCD instrument [33]. Variable-temperature variable-field data were collected at 1.7, 4.13, 6, 10, 20 and 50 K at fields between 0 and 5 T at 615 nm.
Catecholase assays
Catecholase activity was measured against 3,5-di-tert-butylcatechol (3,5-DTBC). Kinetic assays were conducted in acetonitrile (80% saturated with 1 atm O2) at 298 K and formation of product was monitored at 410 nm (ε = 1,900 M−1 cm−1) [19]. Under these conditions no formation of quinone was observed in the absence of the copper complex. In substrate-dependent measurements, the concentration of 1 was held constant at 2.5 × 10−5 M, and the concentration of 3,5-DTBC was varied between 2.5 and 25 mM. For measurement of complex dependence, the concentration of 3,5-DTBC was held constant at 25 mM, and the concentration of 1 was varied between 12.5 and 62.5 μM.
Results
X-ray crystal structure of 1
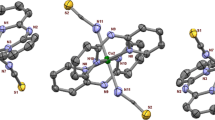
An ORTEP representation of the crystal structure of 1 (Table 1) is shown in Fig. 2, with selected bond lengths and angles given in Table 2. The crystal structure reveals a twofold symmetry axis, such that only half a molecular unit is crystallographically unique. Each copper atom is bound by three nitrogen atoms, the tertiary amine and two pyridines, and by two oxygen atoms from the phenoxo bridge and the terminal acetate. The geometry of the copper centers is that of a distorted square pyramid, with the apical position occupied by the bridging phenoxo oxygen. The degree of distortion from square pyramidal towards trigonal bipyramidal can be quantified using the parameter τ, where τ = (β − α)/60, and α and β are the two basal angles [34]. In an ideal square pyramidal complex τ = 0, while in an ideal trigonal bipyramid τ = 1. Applying this method to 1 gives τ = 0.157, indicating a small deviation from square pyramidal geometry. The square plane is defined by N1, N2, N3 and O2 with the copper atom 0.187 Å out of the plane towards the apical O1. The copper–ligand distances in the square plane are very similar, with a mean value of 1.99 Å, typical of equatorial bond lengths in similar complexes [19, 20, 35, 36]. The distance between the copper and the bridging phenoxo in the axial position is the longest of the copper–ligand bonds, at 2.173 Å. The Cu–Cu distance is 3.916 Å and the Cu–O–Cu′ angle is 128.65°. This metal–metal distance is among the longer distances observed in copper dimers, and similar to that observed in other singly bridged five-coordinate binuclear complexes [19, 21, 35–38]. The bridging phenolate ring is twisted relative to the Cu–O–Cu′ plane with a torsion angle of 46.7°. The solvent water is hydrogen-bonded to the terminally bound acetate (Table S1). Overall, the structure is similar to that reported for the dicopper complex of the BPMP− ligand with terminally bound water molecules [19].

a ORTEP plot of [Cu2(BPMP)(OAc)2][ClO4]·H2O (1), where H-BPMP is 2,6-bis[bis(pyridin-2-ylmethylamino)methyl]-4-methylphenol. Thermal ellipsoids shown at 40% probability. b1 with the first coordination sphere labeled
IR spectroscopy
The IR spectrum of the solid complex indicates a number of ligand stretches in the 1,610–1,400-cm−1 range (1,427, 1,442, 1,473, 1,486, 1,609 cm−1) [39, 40]. The asymmetric and symmetric carboxylate bands are prominent at 1,583 and 1,395 cm−1, respectively. The bands are broad and intense and differ in energy by 188 cm−1, which is at the borderline of the values expected for bridging bidentate and monodentate coordination [41]. This value is consistent with the observation of a hydrogen bond between the terminally bound acetate and the solvent water molecule [41], as observed in the crystal structure. The additional strong peak at 1,078 cm−1 is attributed to the perchlorate counterion.
Visible spectroscopy
In acetonitrile the main features are at 414 nm (ε = 917 M−1 cm−1) and 704 nm (ε = 136 M−1 cm−1), corresponding to the ligand-to-metal charge transfer (LMCT) transition between the bridging phenoxo and the copper, and the d–d transitions, respectively. In methanol/ethanol the bands are blueshifted, such that the LMCT transition is obscured by the intraligand charge transfer transitions, and the d–d band occurs at 642 nm (ε = 243 M−1 cm−1). The spectra are consistent with the square pyramidal geometry about the copper centers determined crystallographically.
EPR spectroscopy
The anisotropic EPR spectrum shown in Fig. 3 is typical of a copper dimer system with dipole–dipole coupling between the two centers. In addition to the allowed transitions around g = 2, the formally forbidden ΔM s = ±2 transitions are also observed around g = 4.3. Computer simulation of the experimental spectrum (Fig. 3a, red) with an axially symmetric spin Hamiltonian (Eq. 1) and the spin Hamiltonian parameters listed in Table 3 yields the spectrum shown in Fig. 3b (blue).

Electron paramagnetic resonance spectrum of 1 measured at 120 K, v = 9.5399 GHz: a experimental spectrum and b computer simulation. Details of the spectra have been enlarged by ×30 and ×8 in the regions of approximately 150 and approximately 270 mT, respectively
The g and A matrices (g ∥ = 2.235 > g ⊥ = 2.065 and A ∥ = 179 × 10−4 cm−1 > A ⊥ = 15 × 10−4 cm−1) for each Cu(II) center are consistent with a tetragonally elongated square pyramidal geometry around the copper ions. A weak exchange interaction (J ∼ −0.6 cm−1) was required to adequately simulate the spectrum. Although the sign of the coupling constant could not be determined from the EPR spectra, the magnetic susceptibility measurements (see below) indicate that the centers are weakly antiferromagnetically coupled (J < 0). The last term in Eq. 1 describes the anisotropic exchange or dipole–dipole interaction and can be related to the internuclear Cu–Cu distance and their relative orientation [42]. The crystallographic data give an angle of 126° for the Cu–O1–Cu angle, which will be the angle between the molecular z-axes, assuming that the tetragonal axis is coincident with the Cu–O1 direction. The simulations were optimum for a somewhat larger angle of 160°, which would indicate that the structure is more open in solution. The simulation gives a Cu–Cu distance of 4.58 Å, greater than that observed in the crystal structure (3.916 Å), in agreement with an increase in the Cu–O1–Cu angle, although it is noted that this value must be an overestimate as it is greater than the combined Cu–O1 distances of 2.173 Å. It has previously been noted that estimates of Cu(II)–Cu(II) separation from dipole–dipole coupling are typically overestimated owing to the unpaired electron spin being delocalized over the \( {d}_{{x^{{\text{2}}} - y^{{\text{2}}} }} \) orbital rather than at a point position, so \( \langle {\text{1/}}r^{3} \rangle \) is less than the distance between atomic positions [43]. The apparent increase in distance could also indicate some level of flexibility of the complex in solution.
Magnetic susceptibility
SQUID data indicated that the complex behaves as a simple paramagnet, and obeys the Curie–Weiss law (Fig. 4; χ M −1 vs. T, red circles). The χ M T versus T plot (Fig. 4, blue crosses) shows a small decrease at low temperatures and the magnetic susceptibility was numerically calculated on the basis of the spin Hamiltonian given in Eq. 1 without the hyperfine interaction and the parameters g iso = 2.23 and J = −0.6 cm−1 (Fig. 4, blue line). Theoretical curves for J = 0 cm−1 (green) and −2 cm−1 (magenta) are also shown in Fig. 4, which show that the fit is quite sensitive to the value of J in the −2JS 1·S 2 term, but is weakly dependent on the S 1·J·S 2 term. Thus, there is very weak antiferromagnetic coupling between the two copper ions. From the relationship χ m T = Ng 2 β 2/2k, an isotropic g of 2.22 is obtained. The μ eff corresponds to a magnetic moment of 1.93 µB per copper center. Although this value is higher than the theoretical value of 1.73 µB, it is within the range typically observed for copper(II) complexes [44–46]. Solution-state susceptibility measurements in acetonitrile yielded a magnetic moment of 2.63 µB (1.87 µB per copper), in good agreement with the SQUID data.

Magnetic susceptibility data of 1. Experimental data points (circles), χM −1 versus T (fit, J = −0.6 cm−1) and χ M T versus T (plus symbols) [fits, green (J = 0 cm−1), blue (J = −0.6 cm−1) and magenta (J = −2 cm−1)]
MCD spectroscopy
The MCD spectra (Figs. 5, 6) reveal intense transitions, a negative peak centered at 615 nm (16,260 cm−1), and a weaker positive peak at 734 nm (13,625 cm−1). The observation of a high-energy negative band and a lower-energy positive band is typical for d–d bands of five-coordinate copper systems [47]. The ΔA/A ratio of approximately 0.2 at 615 nm is consistent with d–d bands. In the IR region weak negative and positive bands are apparent at approximately 910 nm (10,990 cm−1) and approximately 1,500 nm (6,670 cm−1), respectively (Fig. 5).

The absorption (ε) and magnetic circular dichroism (MCD; Δε) spectra measured at 1.7 K, 5 T. The MCD intensity in the IR region is multiplied by a factor of 10

Variable-temperature MCD of 1 in the visible region. Spectra measured at 5 T at 1.7, 4.13, 6, 10, 20 and 50 K
The variable-temperature variable-field MCD (Fig. 7) data of the complex measured at 615 nm at the indicated temperatures reveal superimposable curves, indicative of an S = 1/2 system. The data could be approximately fitted to the simple Eq. 2 [48], yielding g = 2.15, in good agreement with the EPR and magnetization data:

However, using the formalism of Neese and Solomon [49] and Solomon et al. [50] and fixing the g values to those obtained from the EPR data allows additional information on the polarization of this transition to be determined:
Here l i are the directional cosines, 〈S i 〉 are the spin expectation values and M eff ij are the products of two electric dipole transition moments (see supplementary material). The fit of the variable-temperature variable-field data yields the polarization parameters given in Table 3, indicating that this transition is predominantly xy-polarized. The variable-temperature variable-field fit is insensitive to the small values of J. Unlike the EPR spectra, the weak coupling does not affect the electronic spectra and the interpretation of the MCD spectra can proceed in terms of isolated copper centers.
Ligand-field analysis: local environment of the Cu(II) ion
The [Cu2(BPMP)(OAc)2]+ dimer has C 2 symmetry with the rotational axis bisecting the Cu–O1–Cu angle, making the local environment of the two Cu(II) ions equivalent (Fig. S1). The geometry approximates a square pyramid with the Cu(II) slightly above the plane with equatorial bond lengths of 1.94–2.05 Å and a longer axial bond of 2.173 Å to O1. The departure from strict square pyramidal geometry includes angles from the axial to equatorial ligands which range from 90° to 105° and the highly anisotropic π bonding. However, it is instructive to first consider the copper site from the point of an idealized C 4v symmetry. Using standard techniques [51], one can express the d-orbital energies in terms of the e σ and e π bonding angular overlap model (AOM) parameters. In terms of the angle α between the apical to equatorial ligands, the d-orbital energies are given by
where the orbitals are given C 4v symmetry labels and the e σ , e π and the e σ (a) and e π (a) are the σ and π AOM parameters for the equatorial and axial ligands, respectively [51]. Figure 8 shows how these energy levels split in C 4v symmetry (Fig. 8a) and how they change as a function of α relative to the d 9 ground state with the electron hole in the x 2 − y 2 orbital (Fig. 8b). For α = 90°, the geometry corresponds to a holohedralized D 4h symmetry of a tetragonally elongated octahedron, due to the even nature of the d orbitals. Of note in Eq. 4 and Fig. 8 is that the relative energy of the d-orbital states is moderately insensitive to this distortion for α = 90–100° and that the z 2 state will be shifted to higher energy when one considers that e σ > e σ (a) for the longer bond length of the apical ligand. Importantly, the d orbitals are not mixed by the C 4v ligand field, and the E(xz, yz) state remains degenerate. The spin–orbit coupling splits this state (Fig. 8a, right-hand side) and the double point group (C *4v ). Γ notation is used for the spin–orbit states [52] and the more usual Mulliken symbols for the orbital-only states. The L z S z component of the spin–orbit coupling acts within the degenerate E state, resulting in a large relative MCD (ΔA/A), while the lack of an inversion center in the complex will in turn give rise to a large absolute MCD signal.

a The d9 ligand-field states in C4v symmetry both with (right) and without (left) spin–orbit coupling. The spin–orbit coupling removes all but the Kramers degeneracy. Symmetry labels are for the C4v single and C *4v double point groups, respectively. b The ligand-field states as a function of α in units of e σ , assuming e π =0.2e σ and equal equatorial and axial bonding parameters
Considering initially an approximately D 4h symmetry, one may use the perturbation formula given previously for a tetragonally elongated copper(II) complex [53, 54] for both g values and MCD parameters. Using the first-order expressions for the g values [54] with the experimental g values (Table 3) and taking an average energy of the E(xz,yz) and B 2(xy) states as approximately 15,000 cm−1 results in orbital reduction parameters of k ∥ = 0.70 and k ⊥ = 0.78. These values would imply more covalency in the bond to the apical ligand.
Catecholase activity
Catecholase activity was measured in acetonitrile saturated with oxygen (1 atm). The complex displays moderate catecholase activity (k cat = 15 ± 1.5 min−1; K M = 6.4 ± 1.8 mM) with the activated catechol 3,5-DTBC, as shown in Fig. 9a. The reaction rate is linearly dependent on the concentration of the complex, indicating a first-order dependence on catalyst concentration (Fig. 9b).

Substrate a and complex b dependence of oxidation of 3,5-di-tert-butylcatechol by 1
Discussion
Assignment of the ligand-field spectrum
The MCD spectrum shown in Fig. 6 is dominated by the transition to the ligand-field E state which contains unquenched orbital angular momentum. The spin–orbit split components of this state can be shown to have equal and opposite signed C terms (see supplementary material) which overlap to form a derivative-shaped signal or a temperature-dependent pseudo-A term [47]. This shape with the negative feature to higher energy is characteristic for copper(II) in a square pyramidal geometry, as discussed in the supplementary material. The energy separation of the spin–orbit split states would be expected to approach a limiting value equal to the spin–orbit coupling constant λ (approximately 830 cm−1) in the strong tetragonal field limit [55]. The peak positions of the main negative and positive features are separated by approximately 2,500 cm−1. This larger than expected energy separation is due to a shift in the apparent peak positions due to the cancellation of the opposite signed signals. Simultaneously fitting the low-temperature absorption and MCD spectra to peaks that are parameterized by common peak positions [56] (Fig. S2) gives the energy separation of 1,638 cm−1 in Table 4.
Clearly the splitting of these states cannot be due to spin–orbit coupling alone, with the further splitting being due to the low-symmetry ligand field.
Quantifying the ligand field
The ligand field can be quantified using the four ligand-field assignments given in Table 4 and the atomic positions from the crystal structure. A coordinate system on the metal can be defined in terms of the approximate C 4v symmetry, and more importantly, the ligand reference frames can be defined to reflect the local symmetry about each of the metal–ligand bonds. Each ligating atom has an aromatic/conjugated plane such that e π x ≠ 0, e π y = 0 (except for the saturated N1, where e π x = e π y = 0).
Using common e σ and e π parameters for the O and N ligating atoms, there are then four parameters that one can uniquely determine by the four observed transitions. Using the spin–orbital parameter reduced from its free ion value by k = 0.7 and fitting the e σ /e π parameters by minimizing the difference between the experimental and calculated energy levels from a full matrix diagonalization, we could reduce these energy differences to zero with the parameters given in Table 4. The C *4v irreducible representations are then projected onto the wavefunctions, confirming the assignments. The wavefunctions can also be used to calculate the g values explicitly using the magnetic dipole moment operators (k i l i + g e s i ) and these are also given in Table 4. As expected the principal axes are oriented such that g z is approximately in the direction of O1 (rotated away by 8°) and g x and g y are within the approximate plane defined by the equatorial ligands.
Although the e σ /e π AOM parameters have been found for inequivalent (non-symmetry-related) O and N ligands, several remarks can be made. The values of e σ/π(N) > e σ/π(O) reflect the longer O1 bond length and weaker bond strength. The ratios of e σ /e π are remarkably similar (3.6 and 3.7 for N and O, respectively). These are remarkably reasonable values with the approximation that all N and all O ligands have the same radial (e σ/π) parameters. This demonstrates that the transition energies depend on the (angular) geometry and the implicit planes that define the π-bonding anisotropy provided by the crystal structure as well as on the radial parameters.
Spectroscopic studies
The combined MCD, EPR and magnetic susceptibility information, taken together, confirm that the copper centers in the complex are weakly antiferromagnetically coupled (J = −0.6 cm−1). Weak coupling is expected, given that the bridge between the two metals, the μ-phenoxo, is in the apical position of the square pyramids. The unpaired electron is localized in the \( {d}_{{x^{{\text{2}}} - y^{{\text{2}}} }} \) orbital, the lobes of which are directed to the ligand atoms in the basal plane. Electron overlap with the bridging oxygen is therefore negligible. The consistency of the data in solution and solid states indicates limited changes to the structure occur upon dissolution.
In contrast, the catechol oxidase active site is, similar to tyrosinase and hemocyanin, a type 3 copper center [2, 9, 11, 12, 57]. That is, the copper centers are in nitrogen-rich coordination environments and are strongly antiferromagnetically coupled (and hence EPR-silent at X band). Although 1 and catechol oxidases are thus spectroscopically dissimilar in the resting state, they are both able to activate molecular oxygen to oxidize catechol substrates.
Catechol oxidase activity
3,5-DTBC has been widely employed as a substrate in catecholase model complex studies. Owing to its low redox potential, the substrate is readily oxidized and the bulky substituents prevent further reactions such as ring opening. Kinetic parameters of 1 and selected other catechol oxidase model complexes with 3,5-DTBC and the enzyme itself are reported in Table 5 [15, 58, 59]. Comparison is not straightforward, owing to the use of various solvents, temperatures and degrees of solvent oxygenation. The model complexes are at least 3 orders of magnitude less active than the enzyme.
By comparison with other dicopper catecholase model complexes (Table 5), 1, shows moderate catecholase activity. The K M is rather large, though large K M values seem typical of the faster model complexes, and indeed the enzyme itself. The parameters are quite different from those observed by Belle et al. [20] for complexes with the same and similar ligands; however, given the impact that small changes to the methyl group have on the activity of the complexes, this is not unexpected [20, 60]. The isostructural bisaqua complex does not display catecholase activity, while the corresponding μ-hydroxo complex does (Table 5), and it might be expected that the current complex would be similarly inactive. However, the addition of substrate to the hydroxo-bridged complex results in cleavage of the bridge, evidenced by the development of an EPR signal in the previously EPR-silent complex [60]. The presence of a bridging hydroxo is evidently not necessary for catecholase activity.
The presence of labile groups (such as acetate) which may be displaced by the catechol substrate has, however, been shown to be important. Neves et al. [59] found that addition of excess acetate to an acetate-bridged catecholase model moderately inhibited the reaction rate, implying that the acetate and catecholate compete for a binding site in the complex. In contrast, strongly coordinated ligands such as chloride and bromide are not readily displaced and result in catalytically inert complexes [9]. The terminally bound acetate ligands must therefore, similarly to bridging acetates [59, 61], be sufficiently labile to allow binding of the substrate for oxidation. Additionally, it has been suggested that a hydroxo group is required to remove the second proton of the catechol and allow bidentate binding; this would explain the inactivity of the bisaqua complexes [9, 18, 60]. An acetate ligand would, however, contribute a negatively charged moiety capable of facilitating this process.
The final relevant factor to catalysis is the metal–metal distance. In the enzyme, a metal–metal distance of approximately 3 Å in the resting state is observed, and in a model complex with a bridging catecholate (tetrachlorocatecholate) a similar distance of 3.248 Å [62] has been determined, in contrast with the distance of approximately 3.9 Å observed in the present structure. However, other structures with tetrachlorocatecholate bound have suggested that alternative binding modes are plausible [6, 63]. Specifically, Ackermann et al. [6] have reported structures of three binuclear copper(II) complexes in which the doubly deprotonated substrate is bound to one of the copper centers and has hydrogen-bonding interactions with a water ligand on the other copper center. These complexes show Cu–Cu distances of more than 4 Å, consistent with the crystallographic and EPR metal–metal distances determined for 1.
On the basis of the above, there are two possible mechanisms for the observed activity of 1. In both cases, the acetate ligand is likely to assist in deprotonation of the substrate for binding. The catecholate may then bind in a chelating fashion to one of the copper centers, followed by oxidation. Alternatively, the complex may be sufficiently flexible (the EPR metal–metal distance suggests that there is some flexibility) to allow for a bridging coordination followed by oxidation.
In summary, a dicopper complex of the ligand BPMP− was presented as a model for catechol oxidase. The complex has been characterized structurally, as well as spectroscopically by MCD, magnetic susceptibility and EPR. These techniques indicate that the copper centers are weakly antiferromagnetically coupled (J = −0.6 cm−1), due to the only bridging moiety being in the apical position of each of the square pyramidal copper centers. The MCD shows a strong pseudo-A term signal due to unquenched excited state angular momentum arising from the approximate tetragonal symmetry. We have demonstrated from symmetry arguments that the sign of the MCD is a consequence of this geometry. An in-depth ligand-field analysis of the structure and the spectroscopy has elucidated the electronic structure of the complex. Catecholase activity confirms the relevance of the complex as both a structural and a functional model for the active site of catechol oxidase.
References
Mitić N, Smith SJ, Neves A, Guddat LW, Gahan LR, Schenk G (2006) Chem Rev 106:3338–3363
Solomon EI, Sundaram UM, Machonkin TE (1996) Chem Rev 96:2563–2605
Solomon EI, Chen P, Metz M, Lee S-K, Palmer AE (2001) Angew Chem Int Ed 40:4570–4590
Solomon EI, Sarangi R, Woertink JS, Augustine AJ, Yoon J, Ghosh S (2007) Acc Chem Res 40:581–591
Ackermann J, Buchler S, Meyer F (2007) C R Chim 10:421–432
Ackermann J, Meyer F, Kaifer E, Pritzkow H (2002) Chem Eur J 8:247–257
Anekwe J, Hammerschmidt A, Rompel A, Krebs B (2006) Z Anorg Allg Chem 632:1057–1066
Belle C, Selmeczi K, Torelli S, Pierre J-L (2007) C R Chim 10:271–283
Koval IA, Gamez P, Belle C, Selmeczi K, Reedijk J (2006) Chem Soc Rev 35:814–840
Plenge T, Dillinger R, Santagostini L, Casella L, Tuczek F (2003) Z Anorg Allg Chem 629:2258–2265
Gerdemann C, Eiken C, Krebs B (2002) Acc Chem Res 35:183–191
Eicken C, Zippel F, Buldt-Karentzopoulos K, Krebs B (1998) FEBS Lett 436:293–299
Eicken C, Krebs B, Sacchettini JC (1999) Curr Opin Struct Biol 9:677–683
Klabunde T, Eiken C, Sacchettini JC, Krebs B (1998) Nat Struct Biol 5:1084–1090
Mukherjee J, Mukherjee R (2002) Inorg Chim Acta 337:429–438
Rey NA, Neves A, Bortoluzzi AJ, Pich CT, Terenzi H (2007) Inorg Chem 46:348–350
Koval IA, Selmeczi K, Belle C, Philouze C, Saint-Aman E, Gautier-Luneau I, Schuitema AM, van Vliet M, Gamez P, Roubeau O, Luken M, Krebs B, Lutz M, Spek AL, Pierre J-L, Reedijk J (2006) Chem Eur J 12:6138–6150
Granata A, Monzani E, Casella L (2004) J Biol Inorg Chem 9:903–913
Torelli S, Belle C, Gautier-Luneau I, Pierre J-L (2000) Inorg Chem 39:3526–3536
Belle C, Beguin C, Gautier-Luneau I, Hamman S, Philouze C, Pierre J-L, Thomas F, Torelli S (2002) Inorg Chem 41:479–491
Nishida Y, Shimo H, Maehara H, Kida S (1985) J Chem Soc Dalton Trans 1945–1951
Suzuki M, Kanatomi H, Demura Y, Murase I (1984) Bull Chem Soc Jpn 57:1003–1007
Lanznaster M, Neves A, Bortoluzzi AJ, Szpoganicz B, Schwingel E (2002) Inorg Chem 41:5641–5643
Batista SC, Neves A, Bortoluzzi AJ, Vencato I, Peralta RA, Szpoganicz B, Aires VVE, Terenzi H, Severino PC (2003) Inorg Chem Commun 6:1161–1165
Lanznaster M, Neves A, Bortoluzzi AJ, Aires VVE, Szpoganicz B, Terenzi H, Severino PC, Fuller JM, Drew SC, Gahan LR, Hanson GR, Riley MJ, Schenk G (2005) J Biol Inorg Chem 10:319–332
Smith SJ, Casellato A, Hadler KS, Mitić N, Riley MJ, Bortoluzzi AJ, Szpoganicz B, Schenk G, Neves A, Gahan LR (2007) J Biol Inorg Chem 12:1207–1220
Schenk G, Peralta RA, Batista SC, Bortoluzzi AJ, Szpoganicz B, Dick A, Herrald P, Hanson GR, Szilagyi RK, Riley MJ, Gahan LR, Neves A (2008) J Biol Inorg Chem 13:139–155
Kahn O (1993) Molecular magnetism. VCH, New York
Farrugia LJ (1999) J Appl Crystallogr 32:837–838
Altomare A, Cascarano G, Giacovazzo C, Guagliardi A (1993) J Appl Crystallogr 26:343–350
Sheldrick GM (1997) SHELXL97: program for the refinement of crystal structures. University of Gottingen, Germany
Farrugia LJ (1999) J Appl Crystallogr 30:565
Riley MJ, Krausz ER, Stanco A (2003) J Inorg Biochem 96:217
Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC (1984) J Chem Soc Dalton Trans 1349–1356
Oberhausen KJ, Richardson JF, Buchanan RM, McCusker JK, Hendrickson DN, Latour J-M (1991) Inorg Chem 30:1357–1365
Holz RC, Bradshaw JM, Bennett B (1998) Inorg Chem 37:1219–1225
Holz RC, Brink JM, Gobena ET, O’Connor CJ (1994) Inorg Chem 33:6086–6092
Michel F, Torelli S, Thomas F, Duboc C, Philouze C, Belle C, Hamman S, Saint-Aman E, Pierre J-L (2005) Angew Chem Int Ed 44:438–441
Eilers G, Zettersten C, Nyholm L, Hammarstrom L, Lomoth R (2005) Dalton Trans 1033–1041
Lomoth R, Magnuson A, Xu Y, Sun L (2003) J Phys Chem A 107:4373–4380
Deacon GB, Phillips RJ (1980) Coord Chem Rev 33:227–250
Abragam A, Bleaney B (1970) Electron paramagnetic resonance of transition metal ions. Dover, New York
Hitchman MA, McDonald RG, Riley MJ (1984) Inorg Chem 23:2359–2361
Raman N, Ravichandran S, Thangaraja C (2004) J Chem Sci 116:215–219
Chandra S, Sangeetika X (2004) Spectrochim Acta Part A 60:147–153
Hamilton GJ, Kokot E (1972) Aust J Chem 25:2235–2237
Landrum GA, Ekberg CA, Whittaker JW (1995) Biophys J 69:674–689
Johnson MK (2000) In: Que L Jr (ed) Physical methods in bioinorganic chemistry. University Science Books, Sausalito, pp 23–286
Neese F, Solomon EI (1999) Inorg Chem 38:1847
Solomon EI, Neidig ML, Schenk G (2003) In: Lever ABP (ed) Comprehensive coordination chemistry II. Elsevier, Amsterdam, pp 339–349
Schaeffer CE (1968) Struct Bonding 5:68
Koster GF, Dimmock JO, Wheeler RG, Statz H (1963) Properties of the thirty-two point groups. MIT, Cambridge
Dubicki L, Riley MJ, Krausz ER (1994) J Chem Phys 101:1930–1938
Riley MJ, Dubicki L, Moran G, Krausz ER, Yamada I (1990) Inorg Chem 29:1614–1626
Finnie K, Dubicki L, Krausz ER, Riley MJ (1990) Inorg Chem 29:3908–3910
Riley MJ (2007) MCDfit—multiple curve deconvolution and fitting. http://mcdfit.sourceforge.net
Rompel A, Fischer H, Meiwes D, Buldt-Karentzopoulos K, Dillinger R, Tuczek F, Witzel H, Krebs B (1999) J Biol Inorg Chem 4:56–63
Reim J, Krebs B (1997) J Chem Soc Dalton Trans 3793–3804
Neves A, Rossi LM, Bortoluzzi AJ, Szpoganicz B, Wiezbicki C, Schwingel E, Haase W, Ostrovsky S (2002) Inorg Chem 41:1788
Torelli S, Belle C, Hamman S, Pierre J-L, Saint-Aman E (2002) Inorg Chem 41:3983–3989
Gentschev P, Luken M, Moller N, Rompel A, Krebs B (2001) Inorg Chem Commun 4:753–756
Karlin KD, Gultneh Y, Nicholson T, Zubieta J (1985) Inorg Chem 24:3725–3727
Borzel H, Comba P, Pritzkow H (2001) Chem Commun 97–98
Acknowledgments
This work was funded by a grant from the Australian Research Council (DP0558652). The assistance of P.V. Bernhardt with solving the disorder in the crystal structure is kindly acknowledged. The magnetic susceptibility measurements were performed by K.S. Murray and B. Moubaraki.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Smith, S.J., Noble, C.J., Palmer, R.C. et al. Structural and spectroscopic studies of a model for catechol oxidase. J Biol Inorg Chem 13, 499–510 (2008). https://doi.org/10.1007/s00775-007-0334-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-007-0334-7