
Abstract
A systematic in silico approach has been employed to generate sound, experimentally validated active-site models for galactose oxidase (GO) using a hybrid density functional, B(38HF)P86. GO displays three distinct oxidation states: oxidized [Cu(II)–Y•]; semireduced [Cu(II)–Y]; and reduced [Cu(I)–Y]. Only the [Cu(II)–Y•] and the [Cu(I)–Y] states are assumed to be involved in the catalytic cycle, but their structures have not yet been determined. We have developed several models (1–7) for the [Cu(II)–Y•] state that were evaluated by comparison of our computational results with experimental data. An extended model system (6) that includes solvent molecules and second coordination sphere residues (R330, Y405, and W290) is essential to obtain an experimentally correct electronic structure of the active site. The optimized structure of 6 resulted in a five-coordinate Cu site with a protein radical centered on the Tyr–Cys cofactor. We further validated our converged model with the largest model (7) that included additional outer-sphere residues (Q406, H334, Y329, G513, and T580) and water molecules. Adding these residues did not affect significantly the active site’s electronic and geometric structures. Using both 6 and 7, we explored the redox dependence of the active-site structure. We obtained four- and three-coordinate Cu sites for [Cu(II)–Y] and [Cu(I)–Y] states, respectively, that corroborate well with the experimental data. The relative energies of these states were validated by a comparison with experimental redox potentials. Collectively, our computational GO models well reproduce the physicochemical characteristics of the individual states, including their redox behaviors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Galactose oxidase (GO) is an extracellular, monomeric copper protein of molecular mass approximately 68 kDa, isolated from the fungus Dactylium dendroides [1]. It is composed of three distinct domains, organized in an exclusively β-sheet structure with short turns. One domain contains a two-electron redox unit with a mononuclear copper ion and a cross-linked Tyr–Cys radical cofactor that are responsible for its catalytic activity. GO catalyzes the two-electron oxidation of a broad range of primary alcohols (e.g., d-galactose, d-galactoseamine, dihydroxyacetone) to the corresponding aldehydes, concomitantly reducing dioxygen to hydrogen peroxide [1]. GO displays three distinct oxidation states: oxidized ([Cu(II)–Y•], green); semireduced ([Cu(II)–Y], blue); and reduced ([Cu(I)–Y], colorless). The oxidized and the reduced states are catalytically important in the two-electron redox reaction during the catalytic cycle [2].
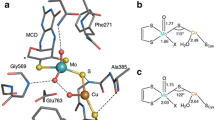
Crystal structures are available for the [Cu(II)–Y] state at pH 4.5 and 7.0 (1.7- and 1.9-Å resolution, respectively). Both structures suggest a distorted square-pyramidal Cu(II) coordination geometry with an axial Y495 ligand and four equatorial ligands of H496, H581, H2O (pH 7.0), or acetate (pH 4.5), and the unique cross-linked Tyr–Cys cofactor (Fig. 1) [3]. Detailed, atomic-resolution structures of both catalytically important states ([Cu(II)-Y•] and [Cu(I)–Y]) are not yet available owing to their inherent instability [4]; however, structural characterization of these redox states is essential to understand the molecular mechanism of GO.

Crystal structure of the active site of galactose oxidase (GO) at 1.9 Å resolution in the [Cu(II)–Y] state including residues known to affect catalytic activity with selected Cu–ligand distances (Protein Data Bank ID 1GOG) [3]. The average Cu–N and Cu–O extended X-ray absorption fine structure (EXAFS) distances for oxidized [Cu(II)-Y•] are 1.97 and 1.96 Å, respectively [5]
Spectroscopic studies have provided considerable insights into the copper coordination environment in GO [2, 5]. On the basis of extended X-ray absorption fine structure (EXAFS) studies, it was suggested that the coordination environment of the oxidized [Cu(II)–Y•] is similar to that of semireduced [Cu(II)–Y] state (Fig. 1) [5]. A three-coordinate Cu site is suggested for the reduced [Cu(I)–Y] state with an average of two Cu–N bond distances and a Cu–O bond distance of 1.95 and 1.97 Å, respectively [6]. In contrast to the geometric structures, the electronic absorption spectra [7] of the GO redox states differ significantly. The [Cu(II)–Y] state shows weak absorption bands at 450 and 620 nm, whereas the [Cu(II)–Y•] state displays multiple, intense electronic transitions that span the visible/near-infrared regions with maximum intensity at 445 and 800 nm. These bands are assigned as ligand-to-metal charge transfer (LMCT) and ligand-to-ligand charge transfer (LLCT) transitions, respectively [4, 8]. The fully reduced [Cu(I)–Y] state shows no characteristic signatures in the electronic spectral region owing to the filled d orbitals of the Cu(I) ion and the nonradical form of the Tyr–Cys cofactor.
A computational study on the catalytic cycle of GO has been previously reported employing a semiquantitative analysis of the experimental data and relevant model systems of GO [9]. More recently, quantum mechanical/molecular mechanics Car–Parrinello simulations of GO and its synthetic active-site analogs were carried out [10] using a small computational model and hybrid density functional theory (DFT; B3LYP) [11]. These results provided new perspectives for developing structure–reactivity correlations for GO. This work suggested a distorted tetragonal Cu site for the [Cu(II)–Y•] state with longer Cu–O (H2O and Y495) distances of 2.6–3 Å, than observed in the EXAFS (1.96 Å) [5, 10]. Notably, these calculations indicated that the protein radical was located on the axial ligand Y495 rather than the equatorial Tyr–Cys cofactor [11]. However, electron paramagnetic resonance studies have demonstrated that the protein radical is on the cross-linked Tyr–Cys cofactor for [Cu(II)–Y•] [12–15]. In addition, the magnetic susceptibility study revealed that the protein radical is antiferromagnetically coupled with the Cu(II) ion, resulting in a diamagnetic ground state for [Cu(II)–Y•] [2]. Therefore, further investigations are needed to reconcile the experimental findings with the calculated electronic and geometric structures of the active site. Moreover, there is a growing interest in designing synthetic biomimetic analogs [16–22] based on the GO active-site architecture for stereoselective alcohol oxidation that can be aided by gaining more insights into the structures of the important oxidation states of GO, and the factors that govern its reactivity.
We describe here a systematic in silico approach for developing accurate active-site models of GO to study the coordination geometries and electronic structures of all three distinct redox states. A spectroscopically calibrated hybrid density functional [B(38HF)P86] was employed, which has been validated for mononuclear copper proteins [23, 24]. Our model-building strategy was designed to evaluate the importance of each inner-shell and outer-shell residue on the geometric and electronic structures of the active site. Each model (Fig. 2) of the oxidized GO was rigorously evaluated by comparing the calculated and experimental structures, spin-density distribution, singlet–triplet energy gap, and electronic absorption spectra. The calculated [Cu(II)–Y] and [Cu(I)–Y] states were obtained by one-electron reduction of the optimized [Cu(II)–Y•] and [Cu(II)–Y] states, respectively. In addition to using the molecular orbital coefficients of [Cu(II)–Y•], [Cu(II)–Y] and [Cu(I)–Y] models as initial orbital guesses for [Cu(II)–Y] (blue solid), [Cu(II)–Y•] (green dotted arrow) and [Cu(I)–Y] (black solid arrow), [Cu(II)–Y] (blue dotted arrow), respectively, we carried out optimizations starting from the crystal structure of the [Cu(II)–Y] form with the ionic fragments [25] (Fig. 3). These fragments correspond to the valence-bond description of amino acid ligands and the Cu(II) ion. These procedures were performed to evaluate the robustness of the optimization method and to investigate protein-strain effects.

Computational models of GO. Changes between two consecutive models are shown in red

Systematic mapping of the potential energy surface of redox states for the GO active site
Overall, our study quantitatively shows that the protein environment has significant influences on the properties of the active site, consistent with previous site-directed mutagenesis studies of second coordination sphere residues (W290H, W290F, W290G, and Y495F) [26, 27]. It is interesting to note that our computational models were developed without following the site-directed mutagenesis study, rather allowing for the model to reveal the residues that are essential in determining the active-site structure. We have already communicated the results of a converged model for the structure of oxidized [Cu(II)–Y•] GO [28]. In this paper, we provide the full details of our model-building strategy for oxidized [Cu(II)–Y•] GO, the semireduced [Cu(II)–Y], and the reduced [Cu(I)–Y] states (Fig. 3). We further validated these models by comparing the redox potentials for [Cu(II)–Y•]/[Cu(II)–Y] and [Cu(II)–Y]/[Cu(I)–Y] with the experimental values [29]. Our work lays a strong foundation for investigating the molecular mechanism of GO. It also provides a demonstrative example for using a systematic in silico model-building approach for constructing structurally and functionally accurate computational models of metalloprotein active sites.
Computational details
A spectroscopically calibrated, hybrid density functional [B(38HF)P86] [24] was employed in our computational study using the Gaussian03 package [30] on a cluster of Intel Xeon EM64T servers. The Becke88 exchange and Perdew86 correlation functional were used with 38% of the total density functional exchange replaced with Hartree–Fock exchange, giving an accurate bonding description for mononuclear copper proteins [23]. The triple-ζ (VTZ*) [31] and double-ζ with polarization [6-31G(d)] [32–34] Gaussian-type all-electron basis sets were employed in all calculations, which form a converged basis set for Cu(II)-containing systems [24, 35].
The generalized ionic fragment approach [25] was employed for model building. First, the molecular orbitals of each residue as ionic ligand fragments were calculated in their crystallographic positions in the [Cu(II)–Y] form (Protein Data Bank ID 1GOG) [3]. Second, these molecular orbitals were combined to give a specific spin-polarized wave function for the complete active-site models. Third, geometry optimizations were performed using spin-unrestricted B(38HF)P86 to maintain the open-shell singlet character of the wave function. As summarized in Fig. 2, our computational models span from a truncated model (H496 and H581 modeled as imidazole, Y495 modeled as phenol, Y272 modeled as thiophenol and H2O) to an extended model system with 214 atoms, which includes the α-carbon of each residue coordinated to the Cu(II); the protein backbone between Y495 and H496; an additional H2O (HOH294) within hydrogen-bonding distance from both Tyr residues (approximately 3 Å in the crystal structure); distal residues Y405, Q406, R330, W290, G513, T580, H334, and Y329, and additional crystal water molecules.
The His residues were modeled as charge-neutral, Y495 as tyrosinate (negatively charged), Y272 as neutral tyrosyl radical, Y405 as protonated Tyr (neutral), R330 as protonated guanidium (positively charged), and all other residues as charge-neutral unless otherwise noted. The total charges of models 1–3 were +1, 0, and −1 and those of 4–7 were +2, +1, and 0 for [Cu(II)–Y•], [Cu(II)–Y], and [Cu(I)–Y], respectively. For models 2–7, α-carbons of each residue were fixed at their crystallographically determined positions during optimization unless otherwise noted. Geometry optimizations were performed using the redundant coordinate system and Berny optimization algorithm [36]. The convergence criteria were set to 0.002 hartree/(au or rad) and 0.01 au or radian RMS change in energy gradients and internal coordinate displacements, respectively. Owing to the relatively lax convergence criteria, geometry optimizations were terminated only when stationary points were firmly located in consecutive calculations. For larger models, this procedure avoided trapping in a higher-energy local minimum. Atomic spin densities were derived from Mulliken population analysis.
The relative energies of Kohn–Sham orbitals from DFT calculation have been shown to provide reasonable estimates of absorption bands for copper proteins [37]. Time-dependent DFT (TD-DFT) [38] calculations were also performed to obtain an improved description of electronic transitions for these models. Antiferromagnetic coupling magnitudes were determined from the energy difference between the optimized singlet and triplet states for each model using spin Hamiltonian J·S A S B (E T − E S = 2J·S Cu(II)·S Tyr•−Cys). Spin projection of the broken-symmetry DFT solution to obtain E S was done by the method suggested by Davidson and Clark [39].
Redox potentials were calculated from the sum of the ionization potentials and solvent reorganization energies relative to the normal hydrogen electrode at −4.43 eV. The solvent reorganization energies were implicitly considered by polarizable continuum calculations using various dielectric constants and solvent radii of 1.385 Å (water). For the proton source in the protonated structures, we used the hydronium ion surrounded by four explicit water molecules. This was further embedded into a standard water-based polarizable continuum. In order to obtain accurate energy for the proton source in the latter system, we used a triple-ζ-quality basis set with both polarization and diffuse functions (6-311+G*).
Results and discussion
First, we demonstrate the importance of using a systematic model-building procedure on the oxidized form of GO, in order to capture all the significant first and second coordination sphere effects. We selected the oxidized state of GO owing to the availability of numerous spectroscopic measurements that are utilized to validate our computational models. Starting from the simplest active-site model (1), we systematically extend this to the converged structural model (6) that reproduces most of the experimental data. We show that a considerably larger model (7) does not improve the calculated electronic and geometric structural data in comparison with experiment relative to model 6. Using models 6 and 7, we describe the structural changes that accompany the relevant redox reactions, and for 6 we also explicitly evaluate the redox potentials.
Systematic development of a converged structural model for oxidized GO
Model 1
The smallest GO model (1) in our computational studies comprises four equatorial ligands, H496, H581, the crosslinked Tyr–Cys cofactor, and water, and an axial ligand, Y495. Both His residues are modeled as imidazoles (charge-neutral), the axial Y495 as phenolate (negatively charged), and the cross-linked thioether–substituted Tyr as a CH3S–substituted phenyl radical. This model is similar to that used in an earlier computational study [11]. The optimized structure of oxidized 1 with selected Cu–ligand distances is shown in Fig. 4a. Model 1 can be characterized as a slightly distorted octahedral Cu site with a phenol ligand in the axial position at 2.29 Å distance. The sulfur atom of the equatorial CH3S–substituted phenyl is located axially, trans to the phenol ligand at a distance of 2.97 Å. Two imidazoles, the substituted phenolate, and the H2O are forming the equatorial plane with Cu–ligand distances ranging from 1.91 to 2.10 Å, and with cis and trans ligand–Cu–ligand angles of 79–100° and 165 and 179°, respectively (Fig. 4a). The aromatic ring of the axial phenol is rotated more than 90° from it is crystallographic position, which suggests the importance of steric constraints from the protein backbone in positioning the aromatic ring almost perpendicular to the equatorial plane (Fig. 1).

Optimized structure of model 1. a Cu–ligand distances (angstroms) and ligand–Cu–ligand angles (degrees). b Spin-density plot (isosurface of ±0.003) and fragment spin densities, and c Kohn–Sham molecular orbital energies (top) and the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) (isosurface of ±0.05) of both α and β manifolds (bottom)
The electronic structure of oxidized 1 (Fig. 4b) reveals 0.74 e − spin density located on the Cu ion, and the rest of the α-spin is delocalized onto the equatorial ligands: both His ligands (0.06 e − on each εN), phenolate (0.09 e −), and H2O (0.04 e −). The β-spin is mainly confined to the axial phenol ring (Fig. 4b), which is inconsistent with experimental findings [12–15]. Since our result agrees with previous computational studies obtained employing a different density functional [10, 11], the location of the spin in this minimal model is likely determined by the computational model and not by the selected functional or basis set.
We have extensively analyzed various initial triplet and singlet state structures of 1 by using a generalized ionic fragment approach [25] with unpaired electrons located at different fragments, such as Cu(II)/substituted phenolate, Cu(II)/axial phenol, and substituted phenolate/axial phenol with a Cu(I) center in the last case (see “Computational details”). In all cases, the structures converged to the same spin-density distribution as described above. Furthermore, the calculated antiferromagnetic coupling (J ∼ 100 cm−1) is considerably smaller than the experimental antiferromagnetic coupling (J > 200 cm−1). In addition, the calculated electronic transitions from a Kohn–Sham orbital analysis are as follows: 266 nm, substituted phenolate to Cu(II) \( {\text{3}}d_{{x^{{\text{2}}} - y^{{\text{2}}} }} {\text{ }} \) (π → σ*), and 469 nm, substituted phenolate to phenyl radical (π → π) (Fig. 4c), which are also in disagreement with the experimental electronic transitions observed for [Cu(II)–Y•] GO. These results obtained for the minimal coordination chemistry model of the active site suggest that the protein backbone between Y495 and H496 and interactions among amino acid side chains are important in constraining the positions of Cu(II) ligands.
Model 2
Model 1 was extended by adding the backbone linkage between Y495 and H496 and the α-carbons of each residue to take into account steric constraints from the protein backbone. Optimization of 2 was performed by freezing all α-carbons at their crystallographic positions. This resulted in a tetragonal copper center similar to in 1, but the axial Y495 and C228 ligands moved away considerably from the copper center (Cu–O(Y495) distance 3.47 Å, and Cu–S(C228) distance 3.56 Å; Fig. 5a). Also, the position of the equatorial H2O is slightly above the equatorial plane and the H2O forms a hydrogen-bonding interaction with the oxygen atom of the axial Y495 (O–O distance 2.67 Å). The average bond distances of 1.98 Å (Cu–N distance 2.00 Å and Cu–O distance 1.94 Å) in 2 are in reasonable agreement with the distances obtained from EXAFS study (Cu–N distance 1.97 Å and Cu–O distance 1.96 Å).

Optimized structure of model 2. a Cu–ligand distances (angstroms) and ligand–Cu–ligand bond angles (degrees). b Spin-density plot (isosurface of ±0.003) and fragment spin densities
The calculated electronic structure of 2 suggests a slightly more covalent bonding than in 1 involving a \( {\text{3}}d_{{x^{{\text{2}}} - y^{2} }} \) orbital with 0.67 e − α-spin density located on the Cu(II) ion, and the remaining 0.34 e − spin density delocalized onto the σ-bonded equatorial ligands [Y272 (0.15 e −), H496 (0.06 e −), H581 (0.08 e −), and H2O (0.05 e −)]. The protein radical (β-spin) is mainly localized onto a π orbital of axial Y495 (Fig. 5b). The calculated antiferromagnetic coupling J (approximately 7 cm−1) is almost negligible, yielding nearly degenerate triplet and singlet states. Hence, the calculated electronic structure of 2 is inconsistent with the experimental data in the location of protein radical, the negligible singlet–triplet energy gap, and the inaccurate electronic transitions (Fig. S1).
Model 3
We reasoned that for changing the location of the Tyr radical from axial to equatorial it would be desirable to reduce the nucleophilicity of the axial Tyr (Y495) ligand and thus destabilize a radical at this position. Accordingly, model 2 was extended via addition of a hydrogen-bonding water molecule, which is resolved in the crystal structure [3]. The optimized 3 with the constraints as for 2 resulted in a tetragonal pyramidal copper site with all equatorial ligands located within 1.89–2.0 Å distances; the shortest being the Cu–O(Y272) (Fig. 6a). The axial Y495 is located at 2.49 Å from the copper center, and the Tyr ring is almost parallel to the equatorial plane. The electronic structure of 3 shows small changes in the electron spin distribution despite the geometric difference relative to 2, and results in a triplet ground state (Fig. 6b).

Optimized structure of model 3. a Cu–ligand distances (angstroms) and ligand–Cu–ligand angles (degrees). b Spin-density plot (isosurface of ±0.003) and c spin-density plot of Y495 protonated in [Cu(II)–Y•] model 3 (isosurface of ±0.003)
In order to further probe the idea that the reduced nucleophilicity of axial Y495 could tune the location of the protein radical, we evaluated the possibility of the O(Y495) being protonated. It is worth noting that Himo et al. [11] also reported that in the substrate-bound form of oxidized GO the radical is transferred to the equatorial Tyr–Cys cross-link simultaneously with the proton transfer to O(Y495) from the substrate. In our case without substrate present, a single point energy calculation for the protonated 3 resulted in a spin distribution that is consistent with experiment. For the first time, the radical (unpaired β-spin electron) is located on an out-of-plane π orbital of the Tyr–Cys cofactor and the unpaired α-spin electron is localized on the Cu \( {\text{3}}d_{{x^{{\text{2}}} - y^{2} }} \) orbital and the in-plane orbitals of the covalently bound equatorial ligands (Fig. 6c). This result motivated us to assess the possibilities of other hydrogen-bonding residues around the oxygen atom of Y495, leading to model 4.
Model 4
The oxygen atom of the Tyr (Y405) residue is located at approximately 3.25 Å away from the O(Y495) and it is well positioned for hydrogen bonding with Y495. Furthermore, a positively charged Arg residue (R330) is located within hydrogen-bonding range of H2O (294), which could stabilize the position of this water molecule. Note that the position of H2O (294) in model 3 changed considerably by moving toward Y272 (d O–O(Y272) = 2.66 Å) and forming a stronger hydrogen-bonding interaction with Cu-coordinated H2O (d O–O = 2.58 Å) relative to the crystal structure (d O–O(Y272) = 3.02 Å and d O–O = 3.01 Å). Therefore, model 4 consists of protonated second coordination sphere residues Y405 and R330 in addition to the residues included in 3 (Fig. 2). Initially, geometric optimization of 4 was performed with fixed crystallographic positions for α-carbons of each residue (4a). As shown in Fig. 7a, model 4a has a tetragonally distorted pyramidal coordination geometry with H2O in an axial position at 2.49 Å, whereas Y495 now occupies an equatorial position. The equatorial plane is defined by the four shortest Cu–ligand distances (1.94–2.06 Å), with cis and trans ligand–Cu–ligand bond angles of 91–105° and 114 and 153°, respectively (Fig. 7a). With respect to the electronic structure, this means that the equatorial plane containing the \( {\text{3}}d_{{{\text{x}}^{{\text{2}}} - y^{2} }} \) orbital is rotated by 90° relative to that in models 1–3.

Optimized structure of model 4a. a Cu–ligand distances (angstroms, solid lines) and hydrogen-bonding interaction (angstroms, dotted lines) and ligand–Cu–ligand angles (degrees). b Spin-density plot (isosurface of ±0.003)
The calculated electronic structure of the oxidized [Cu(II)–Y•] state of 4 suggests a metal-centered bonding description involving a \( {\text{3}}d_{{x^{{\text{2}}} - y^{2} }} \) orbital with 0.76 e − α-spin density located on the Cu(II) ion, and the remaining 0.24 e − is delocalized evenly onto the σ-bonded equatorial ligands (Fig. 7b). The protein radical (Tyr•–Cys) has approximately 0.94 e − β-spin density in an out-of-plane π orbital, including approximately 0.20 e − spin delocalization onto the sulfur atom of C228. The possible involvement of S orbitals in the protein radical predicted by this model parallels the conclusions from studies of biomimetic GO models [14, 40].
An alternative optimization of oxidized 4 was performed with additional distance constraints from EXAFS [both Cu–N distance 1.97 Å and Cu–O(Y272) distance 1.96 Å] in addition to fixing α-carbons at their crystallographic positions (4b). Interestingly, this approach produced a trigonally distorted tetrahedral Cu(II) geometry with ligand–Cu–ligand angles spanning from 99 to 125° (Fig. 8a), which is quite different from the situation for 4a, and resembles the angular parameters of blue copper proteins [37]. The equatorial H2O in 4b is quite distant from the copper center at 3.42 Å. Both models (4a and 4b) agree with EXAFS results but differ in their electronic structures. In 4b the Cu(II) spin density was increased to approximately 0.86 e − α-spin, corresponding to less covalent bonding (Fig. 8b). The delocalization of the protein radical (Tyr•–Cys) onto the sulfur atom of C228 was also reduced by about 4% (Fig. 8b). The calculated antiferromagnetic coupling constants are 923 and 39 cm−1 for 4a and 4b, respectively. It is also important to note that model 4a is approximately 68 kJ mol−1 lower in energy than 4b; therefore, model 4a was considered a more reasonable model than 4b. TD-DFT analysis was performed for 4a and predicted three intense features at 766, 390, and 372 nm, giving rather close agreement with the experimental optical transition. They can be assigned to tyrosinate (π 3 → π 4), Y272 (π 2 → π 4), and Y272 (π 3) → σ* of Cu(II) \( {\text{3}}d_{{x^{{\text{2}}} - y^{{\text{2}}} }} \) transitions, respectively.

Optimized structure of model 4b. a Cu–ligand distances (angstroms, solid lines), hydrogen-bonding interactions (angstroms, dotted lines), and ligand–Cu–ligand angles (degrees). b Spin-density plot (isosurface of ±0.003)
Model 5
While 4a can already be considered as a reasonable computational model for the [Cu(II)–Y•] state of the GO active site, it lacks a key residue (W290) that has been shown to influence the catalytic activity by affecting the generation of the tyrosyl radical (Y272) in GO [41]. In the crystal structure, it also appears that the conformation of Y272 is affected by a π stacking interaction with W290. This stacking interaction was included in model 5 to evaluate the effect of Trp on the geometric and the electronic structure of the [Cu(II)–Y•] state. The optimized structure of oxidized 5 resulted in a square-planar copper site with all equatorial ligands located within 1.87–2.04 Å distance, the shortest being Cu–O (Y272). The axial Y495 is located 3.17 Å from the copper center, and almost parallel to the equatorial plane of the copper center. Unexpectedly, R330 and Y405 residues were distorted significantly in the optimized structure relative to the crystal structure, in contrast to the results for 4. W290 was also dislocated from the stacking plane. Moreover, the electronic structure of 5 is inconsistent with experiment since the protein radical was mainly localized on a π orbital of the axial Y495 (Fig. S2).
These results for model 5 indicate that the π–π stacking interaction of the cross-linked Tyr–Cys cofactor and W290 can have a significant influence on the electronic structure of the [Cu(II)-Y•] state. We inferred that the protein environment must modulate the interaction between W290 and the Tyr–Cys cofactor. Inspection of the crystal structure revealed that both the axial Y495 and the indole nitrogen of W290 are partially exposed to solvent. Because our computational work already revealed that hydrogen bonding can modulate the electron spin distribution in the GO active site, we introduced an explicit water solvent molecule that is within hydrogen-bonding distance of the indole nitrogen of W290 or of the oxygen of Y495 in separate models. Only the latter hydrogen-bonding interaction resulted in a protein radical on the cross-linked Y272–C228 cofactor defining our model 6.
Model 6
The spin-polarized wave function of 6 was obtained from two different starting points owing to issues in self-consistent-field convergence: merging ionic wave functions from individual fragments; or adding the W290 residue and the water molecule to a covalent, spin-polarized wave function of 4. The latter resulted in a lower-energy wave function (atomic spin densities of 0.74 e − on Cu, −0.60 e − on Y272, −0.15 e −on Y495, and −0.10e − on W290) by approximately 104 kJ mol−1 relative to the former wave function (atomic spin densities of 0.99 e − on Cu, −0.75 e − on Y272, −0.14 e − on Y495, and −0.09e − on W290). It is worth noting that these results demonstrate well potential pitfalls for finding the ground-state electronic state in large computational models from single-point calculations. The generalized ionic fragment approach may avoid these pitfalls by allowing for generating all possible valence-bond structures as initial wave functions in a systematic and reproducible manner. Optimization of 6 starting from the lower-energy spin-polarized wave function was performed with additional constraints of R330 (δN, εC, and χN) in order to avoid the unreasonable distortion of R330 that was observed in 5. The optimized structure of 6 reveals a five-coordinated square-pyramidal Cu coordination geometry with Y495 in an axial position and the other ligands in equatorial positions with cis and trans ligand–Cu–ligand bond angles of 77–94° and 155 and 159°, respectively (Fig. 9a). The overall coordination environment of the copper site in [Cu(II)–Y•] is comparable to that in the crystal structure of the [Cu(II)–Y] state with some deviations in the Cu–ligand bond distances. It is interesting to note that the average Cu–N and Cu–O distances (1.99 and 2.06 Å) also slightly deviate from the corresponding EXAFS values. The calculated electronic structure of the oxidized [Cu(II)-Y•] state of 6 suggests a metal-based bonding description involving a \( {\text{3}}d_{{{\text{x}}^{{\text{2}}} - y^{2} }} \) orbital with 0.76 e − α-spin density located on the Cu(II) ion. The Tyr•–Cys radical has approximately 0.91e − β-spin density in an out-of-plane π 4 orbital (Fig. 9b; for orbital labeling see Fig. S3). This includes approximately 0.24 e − spin delocalization onto the sulfur atom of C228. The π–π stacking between W290 and Tyr•–Cys (approximately 3.7 Å), and constraints from α-carbon atoms of Y272, C228, and W290 maintain a cofactor orientation (Cu–O–C bond angle, α = 125° and dihedral angle between equatorial plane of Cu coordinated ligands and tyrosyl ring plane, β ∼ 82°) that results in a diamagnetic ground state [22].

a Optimized structure of model 6 with Cu–ligand (angstroms, solid lines), hydrogen-bonding (angstroms, dotted lines) distances and ligand–Cu–ligand angles (degrees). b Spin-density plot (isosurface of ±0.003). c Relative energies of Kohn–Sham molecular orbitals (top) and HOMO-1 and LUMO (isosurface of ±0.05) of both α and β manifolds (bottom)
It is important to mention that the proton on the indole N of W290 is bent and about 0.03 e − spin density is located on the Trp ring. A Kohn–Sham molecular orbital energy diagram for the oxidized model is shown in Fig. 9c. The electronic transitions between HOMO-1 and LUMO orbitals at 311 and 601 nm in the spin-down (β) and the spin-up (α) manifolds, respectively, can be assigned to LMCT and LLCT excitations. The former corresponds to the tyrosinate (Y495) to Cu \( {\text{3}}d_{{x^{{\text{2}}} - y^{{\text{2}}} }} \) orbital,
π 4 → σ*, and the latter to tyrosinate (Y495) to tyrosyl (Y272) radical, π 4 → π 4 (Fig. 9c). TD-DFT analysis gave an improved agreement between the experimental and calculated band positions and intensities. Nonzero oscillator strengths (f) were obtained for transitions at 795 nm (f = 0.02, radical Y272 π 3 → π 4), 400 nm (f = 0.04, radical Y272 π 2 → π 4), and 359 nm [f = 0.05, H496 (π) and Y272 (π 4) to σ* of Cu(II) \( {\text{3}}d_{{x^{{\text{2}}} - y^{2} }} \)]. These are consistent with assignments based on electronic absorption and resonance Raman measurements [42, 43] except that the TD-DFT calculations do not predict substantial contributions from Y495, as inferred from the resonance Raman experiments. The calculated antiferromagnetic coupling is about 752 cm−1 for 6.
Model 7
In order to demonstrate that model 6 is indeed a converged structural model for oxidized GO, which already provides good agreement with experiment, we further extended this computational model to 214 atoms. This extended model contains second and third coordination sphere residues: Q406, H334, Y329, T580, G513, and additional H2O that are located within hydrogen-bonding distance of R330, Y405, R330, H581 and H496, respectively (Fig. 2). The optimized structure reveals a five-coordinate square pyramidal geometry highly similar to 6 without significant distortion of any residues (Fig. 10a) relative to their crystallographic positions.

a Optimized structure of [Cu(II)–Y•] model 7 with Cu–ligand (angstroms, solid lines) and hydrogen-bonding (angstroms, dotted lines) distances and ligand–Cu–ligand angles (degrees). b Spin-density plot (isosurface of ±0.003)
The Q406 residue that keeps the guanidium group of R330 in place was replaced by defining constraints for δN, εC, and χN atoms of R330. Importantly, the calculated electronic structure for the [Cu(II)–Y•] state is identical to model 6 as well (Fig. 10b). The antiferromagnetic coupling constant J was only slightly affected (approximately 608 cm−1) with the same preference of the open-shell singlet ground state relative to the triplet state. Thus, model 6 provides a sufficient description of the structure of the [Cu(II)–Y•]. Model 7 may be needed in future investigation of the molecular mechanism of the GO chemical function.
Redox dependence of the active-site structure
While model 6 has been shown to be the converged computational model that reasonably reproduces key experimental data for the [Cu(II)–Y•] state of GO, the redox dependence of the active-site structure required a minor, yet important modification to this model. The change in the total charge of the computational model that accompanies reduction and the concomitant decrease of covalent interactions between the Cu center and its ligands required the introduction of additional geometric constraints to 6 that prevent unreasonable displacements of residues relative to their crystallographic positions. These additional constraints on β- and γ-C atoms of W290 affect the optimized model of the [Cu(II)–Y•] state negligibly. The introduction of these constraints is not arbitrary as they make up for the absence of hydrogen-bonding interactions from the Y329 residue that is present explicitly in model 7.
The potential energy surface of the three redox states were rigorously probed as summarized in Fig. 3. In the following we will only discuss the lowest-energy, final stationary structures obtained for each redox step.
Semireduced state of GO [Cu(II)–Y]
Geometry optimizations from different starting points resulted in three different stationary points that are connected with a distortion coordinate along the Cu–O(Y495) and the Cu–OH2 bonds. This distortion coordinate spans a tetragonal Cu(II) coordination geometry with short Cu–OH2 (2.09 Å) and long Cu–O(Y495) (2.30 Å) distances, a structure with equivalent Cu–O distances (2.16 Å), and the lowest-energy semireduced [Cu(II)–Y] structure, which possesses a long Cu–OH2 (2.24 Å) and a short Cu–O(Y495) (2.05 Å) bond (Table S1). The energy differences among these models are within the accuracy of the selected level of theory (approximately 4.4 kJ mol−1). Going from the oxidized to the semireduced state, we find the most significant change is the shorter Cu–O(Y272) distance (2.02 vs. 1.92 Å) (Figs. 9a, 11). The calculated average Cu–O and Cu–N distances (1.99–2.05 and 2.00–2.05 Å) are slightly longer than the reported EXAFS [5] distances (1.97 and 1.95 Å). Independent of the position of a model structure along the distortion coordinate, the atomic spin densities remain similar: about 0.72–0.75 e − on the Cu(II) ion, with the remaining 0.25 e − distributed onto the equatorial ligands (Y272, 0.10–0.12 e −; H496, H581, 0.04–0.07 e −; H2O, 0.02–0.04 e −). TD-DFT analysis revealed that electronic transitions of these three structures are similar in energy to the experimental absorptions at 468 and 625 nm, which correspond to LMCT [Y272 (π) to σ* of Cu(II) \( {\text{3}}d_{{x^{{\text{2}}} - y^{2} }} \)] and ligand-field transitions, respectively (Table S2).

Optimized structure of [Cu(II)–Y] from [Cu(II)–Y•] 6 with Cu–ligand (angstroms, solid lines) and hydrogen-bonding (angstroms, dotted lines) distances and ligand–Cu–ligand angles (degrees)
The semireduced model (7) also parallels the result obtained from semireduced model 6 (Fig. S4, Table S3); however, the rotation of W290 is observed even in this extended model. Additional constraints (β-carbons of Y272 and W290) were applied to prevent this structural change, which had insignificant influence on the coordination geometry and the electron spin density distribution.
As a further validation of the semireduced computational model, we determined the one-electron redox potential for the [Cu(II)–Y•]/[Cu(II)–Y] couple. The ionization potential of [Cu(II)–Y•]/[Cu(II)–Y] is 7.88 V and by increasing the dielectric constant of the polarizable continuum surrounding model 6, this decreases closer to the experimental value. The calculated redox potential is 691 mV with a dielectric constant of 10, which is a typical dielectric constant for a buried redox-active site (Table S4). However, the GO active site is partially solvent exposed and thus it is not surprising that a higher dielectric constant (ε = 40) gives a better agreement (calculated, 439 mV: experimental, 400 mV) [29]. The good agreement between the experimental and the calculated spectroscopic features, average Cu–O/Cu–N bond lengths, and redox potentials suggests that model 6 captures most of the important structural features and interactions in the protein environment for both the oxidized and the semireduced states of GO.
Reduced state of GO [Cu(I)–Y]
Starting from various initial structures for the reduced state (Fig. 3), we located stationary points along a similar distortion coordinate as described for the semireduced state. These range from a Cu(I) site with a trigonal pyramidal coordination geometry (higher energy) to the almost trigonal planar structure, where the Cu(I) is coordinated by two His N atoms and a Tyr O atom. The average Cu–(N/O) bond distance of 2.01 Å (Fig. 12a, Table S5) is close to the experimental EXAFS distance of 1.99 Å [6]. The π stacking of W290 and the Tyr–Cys cofactor is perturbed as expected from the increased electrostatic repulsion between the two-electron-reduced active site and the electron-rich W290. For model 7 only the trigonal planar structure (Table S6) was localized as a stationary point, with a W290 rotated away from the π-stacking plane owing to rotation along the βC–γC bond in the W290 residue.Footnote 1

Optimized structures of a [Cu(I)–Y], b Y495-protonated [Cu(I)–Y], and c Y272-protonated [Cu(I)–Y] with Cu–ligand (angstroms, solid lines) and hydrogen-bonding (angstroms, dotted lines) distances and ligand–Cu–ligand angles (degrees)
The redox potential calculated using a dielectric constant of 40 for the [Cu(II)–Y]/[Cu(I)–Y] couple is approximately 930 mV more negative than the experimental value of 160 mV [29]. This is a rather large difference and suggests that the reduced form may be protonated. Furthermore, Wright and Sykes [29] have described the pH dependence of the redox potential for the [Cu(II)–Y]/[Cu(I)–Y] couple. In order to investigate this, the reduced structure was protonated at O(Y495) and in a separate model at O(Y272). Both protonated states gave a three-coordinated Cu site (Fig. 12b, c), as also found by X-ray absorption spectroscopy [6], with Cu(I) ions coordinated by two N from His residues and O of tyrosinate, resulting in trigonal pyramidal structures. The redox potentials for the semireduced/Y495-protonated and semireduced/Y272-protonated reduced couples in low dielectric medium (ε = 10) were calculated to be −163 and −40 mV, respectively, which are closer to the experimental value of 160 mV than was the redox potential of the nonprotonated, reduced form. A tetrasolvated hydronium (H3O+) ion was used as the source for the protons with an energetic cost of 11.9 eV.
Adjustment of the dielectric constant towards more polar environments (ε = 40) only slightly affected these redox potentials (−173 and −54 mV for Y495 and Y272 protonated states, respectively). While these potentials are somewhat less accurate than that of the oxidized/semireduced couple, the deviation of 300 mV between the calculated and the experimental potentials is already a rather reasonable agreement for a DFT computational model without the explicit consideration of point charges and dipoles from the rest of the protein environment.
Furthermore, the redox potentials calculated for a low dielectric medium (ε = 10) for the catalytically relevant two-electron redox couples of [Cu(II)–Y•]/[Cu(I)–HY495] and [Cu(II)–Y•]/[Cu(I)–HY272] are 264 and 325 mV, respectively. These values are in close vicinity of the experimental value (approximately 275 mV). In a more polar environment (ε = 40), these calculated redox potentials shift towards less positive values (133 and 193 mV) with a deviation of 80–140 mV from the experimental values.
Conclusion
It has already been well documented experimentally by site-directed mutations that residues of the second coordination shell influence substrate binding and catalysis in GO. Our current computational study parallels these site-directed studies in emphasizing the importance of the contributions from second coordination sphere residues. Furthermore, we would like to put this study forward as an example for evaluating contributions from the protein matrix to the active-site structure and catalytic activity. Inclusion of outer-sphere ligands in the in silico models was crucial to obtain the spectroscopically correct electronic structure for the active site of GO. The hydrogen-bonding interactions involving the O atom of Y495 and the π–π stacking of W290 and the Tyr•–Cys cofactor play a critical role in modulating the spin-density distribution around the Cu site and thus the molecular orbitals involved in catalysis. Since Y495 and W290 are both exposed to water solvent, inclusion of hydrogen-bonding interactions from explicit solvent water molecules with these residues is also essential. Our oxidized [Cu(II)-Y•] models (the smallest required model 6 and an optimal model 7) with a tetragonal square-pyramidal active-site geometry and with a radical centered on the Tyr–Cys cofactor gives good agreement with the experimentally observed optical transitions and magnetic coupling constant. For an overview, the location of the protein radical, magnetic coupling and electronic transitions from all the computational models are provided as supplementary material (Table S7).
Using these models, we have developed the structure of the fully reduced [Cu(I)–Y] state, which is also implicated in the catalytic mechanism, as well as the resting inactive, semireduced [Cu(II)–Y] state. The lowest-energy semireduced and fully reduced states were shown to have distorted tetragonal and trigonal planar Cu sites, respectively. We were able to estimate reasonably well the catalytically relevant redox potentials for the [Cu(II)–Y•]/[Cu(I)–HY] couple (133–193 mV; experimental, approximately 275 mV) in an intermediate dielectric environment with ε = 40. The protonation states of axial Y495 and the equatorial Y272 were found to be critical for obtaining experimentally sound redox potentials. Similarly to X-ray absorption spectroscopy studies on the substrate reduced form, both protonated states show a three-coordinate copper site. Overall model 6 is the smallest converged model for the GO active site which captures all the essential elements for the active site of GO and 7 is sufficiently extended to validate conclusions based on 6. Our computational work sets the stage for a detailed investigation of substrate binding and the catalytic cycle of GO.
Notes
It is important to note that the large number of constraints employed for the optimized structures for both model 6 and model 7 seem to have a negligible effect on the coordination geometry, yet rotation of W290 is observed for the reduced structure owing to the conformation flexibility of the βC–γC bond of W290. Since W290 is partially exposed to the solvent, it is quite likely that the distortion observed in the computational model can occur upon complete reduction of the GO active site in a protein environment.
References
Avigad G, Amaral D, Asensio C, Horecker BL (1962) J Biol Chem 237:2736–2743
Whittaker JW (2003) Chem Rev 103:2347–2363
Ito N, Phillips SEV, Stevens C, Ogel ZB, McPherson MJ, Keen JN, Yadav KDS, Knowles PF (1991) Nature 350:87–90
Whittaker MM, Whittaker JW (1988) J Biol Chem 263:6074–6080
Knowles PF, Brown RD, Koenig SH, Wang S, Scott RA, McGuirl MA, Brown DE, Dooley DM (1995) Inorg Chem 34:3895–3902
Clark K, Penner-Hahn JE, Whittaker M, Whittaker JW (1994) Biochemistry 33:12553–12557
Whittaker JW (2002) Adv Protein Chem 60:1–49
Whittaker MM, Whittaker JW (2000) Protein Expr Purif 20:105–111
Wachter RM, Branchaud BP (1998) Biochim Biophys Acta Protein Struct Mol Enzymol 1384:43–54
Rothlisberger U, Carloni P, Doclo K, Parrinello M (2000) J Biol Inorg Chem 5:236–250
Himo F, Eriksson LA, Maseras F, Siegbahn PEM (2000) J Am Chem Soc 122:8031–8036
Babcock GT, El-Deeb MK, Sandusky PO, Whittaker MM, Whittaker JW (1992) J Am Chem Soc 114:3727–3734
McGlashin ML, Eads DD, Spiro TG, Whittaker JW (1995) J Phys Chem 99:4918–4922
Gerfen GJ, Bellew BF, Griffin RG, Singel DJ, Ekberg CA, Whittaker JW (1996) J Phys Chem 100:16739–16748
Whittaker MM, Chuang YY, Whittaker JW (1993) J Am Chem Soc 115:10029–10035
Halfen JA, Young VG Jr, Tolman WB (1996) Angew Chem Int Ed Engl 35:1687–1690
Halfen JA, Jazdzewski BA, Mahapatra S, Berreau LM, Wilkinson EC, Que L Jr, Tolman WB (1997) J Am Chem Soc 119:8217–8227
Chaudhuri P, Hess M, Weyhermueller T, Wieghardt K (1999) Angew Chem Int Ed Engl 38:1095–1098
Chaudhuri P, Hess M, Mueller J, Hildenbrand K, Bill E, Weyhermueller T, Wieghardt K (1999) J Am Chem Soc 121:9599–9610
Chaudhuri P, Hess M, Flowers L, Wieghardt K (1998) Angew Chem Int Ed Engl 37:2217–2220
Kruse T, Weyhermuller T, Wieghardt K (2002) Inorg Chim Acta 331:81–89
Muller J, Weyhermuller T, Bill E, Hildebrandt P, Ould-Moussa L, Glaser T, Wieghardt K (1998) Angew Chem Int Ed Engl 37:616–619
Szilagyi RK, Solomon EI (2002) Curr Opin Chem Biol 6:250–258
Szilagyi RK, Metz M, Solomon EI (2002) J Phys Chem A 106:2994–3007
Szilagyi RK, Winslow M (2006) J Comput Chem 27:1384–1397
Ito N, Phillips SEV, Yadav KDS, Knowles PF (1994) J Mol Biol 238:794–814
Rogers MS, Knowles PF, Baron AJ, McPherson MJ, Dooley DM (1998) Inorg Chim Acta 275–276:175–181
Rokhsana D, Dooley DM, Szilagyi RK (2006) J Am Chem Soc 128:15550–15551
Wright C, Sykes AG (2001) J Inorg Biochem 85:237–243
Frisch MJ et al (2005) Gaussian 03, revision D 01. Gaussian, Pittsburgh
Schaefer A, Horn H, Alrichs R (1992) J Chem Phys 97:2571–2577
Rassolov VA, Pople JA, Ratner MA, Windus TL (1998) J Chem Phys 1223–1229
Francl MM, Hehre WJ, Binkley JS, Gordon MS, DeFrees DJ, Pople JA (1982) J Chem Phys 77:3654–3665
Hariharan PC, Pople JA (1973) Theor Chim Acta 28:213–222
Ryde U, Olsson MHM, Pierloot K (2001) Theor Comput Chem 9:1–55
Farkas O., Schlegel HB (1998) J Chem Phys 109:7100–7104
Solomon EI, Szilagyi RK, George SD, Basumallick L (2004) Chem Rev 104:419–458
Stratmann RE, Scuseria GE, Lovell T (1998) J Chem Phys 109:8218–8224
Davidson ER, Clark AE (2005) Int J Quantum Chem 103:1–9
Itoh S, Taki M, Kumei H, Takayama S, Nagatomo S, Kitagawa T, Sakurada N, Arakawa R, Fukuzumi S (2000) Inorg Chem 39:3708–3711
Rogers MS, Tyler EM, Akyumani N, Kurtis CR, Spooner RK, Deacon SE, Tamber S, Firbank SJ, Mahmoud K, Knowles PF, Phillips SE, McPherson MJ, Dooley DM (2007) Biochemistry 46:4606–4618
McGlashen ML, Eads DD, Spiro TG, Whittaker JW (1995) J Phys Chem 99:4918–4922
Whittaker MM, DeVito VL, Asher SA, Whittaker JW (1989) J Biol Chem 264:7104–7106
Acknowledgements
We gratefully acknowledge the support of this research by NIH grant GM 27659 for D.M.D. and by the Montana State University Thermal Biology Institute (NASA NNG04GR46G) and the ONR Center for Bioinspired Nanomaterials (N00014-06-01-1016) for R.K.S.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Rokhsana, D., Dooley, D.M. & Szilagyi, R.K. Systematic development of computational models for the catalytic site in galactose oxidase: impact of outer-sphere residues on the geometric and electronic structures. J Biol Inorg Chem 13, 371–383 (2008). https://doi.org/10.1007/s00775-007-0325-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-007-0325-8