
Abstract
Density functional theory calculations have been employed to study the interaction between the Zn2+ ion and some standard amino acid models. The highest affinities towards the Zn2+ ion are predicted for serine, cysteine, and histidine. Relatively high affinities are reported also for proline and glutamate/aspartate residues. It was found that the zinc complexes with cysteine adopt a tetrahedral conformation. Conversely, complexes with one or two histidine moieties remain in an octahedral geometry, while those with three or more histidine groups adopt a square-planar geometry.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Metal ions play an important role in the structure and function of many biomacromolecules. They can be found at active sites of enzymes [1], have an important role in photosynthesis [2], act as secondary messengers [3], promote and stabilize native conformations of nucleic acids [4], and facilitate protein–DNA binding [5]. Metal-based compounds may also be used as potent antibacterial, antifungal, and anticancer drugs [6], or as imaging agents [7]. In all those cases, the metal ion–amino acid and metal ion–nucleic acid interactions play a central role.
Recently, metal ions have been also used in metallization of biomacromolecules [8]. These processes rely upon specific metal ion–amino acid or metal ion–nucleic acid interactions, which allow for an efficient metal deposition and attachment to biological systems. The biomolecule metallization has possible application in the fabrication of nanoscale structures [9]. The molecular mechanism of the metallization process has been studied by means of quantum chemical calculations of metal ion–amino acid interactions [10]. It has been found that high chemical affinities of specific sites within the macromolecule towards the metal ion usually depend on the interplay of at least two amino acid moieties. In some cases, one of the amino acids involved in the metal ion binding may have a rather small affinity towards that ion, but its combination with a second amino acid may result in a combined high metal affinity leading to a strong binding. It has been noted that, in order to describe the metallization process of a biomacromoleule, one needs to know the metal ion affinities towards all amino acids contained in the biomolecule.
Despite the fact that for most metal ions, the amino acid affinity values have been determined experimentally, those values may not be useful in describing the metallization process. The reason for that is the following. Owing to the presence of a carboxyl group (which has a high affinity towards many metal ions) in every amino acid molecule, the metal ion–amino acid interaction usually involves this group. As a result, the experimental values of most of the metal ion–amino acid affinities are similar, with the exception of those amino acids that interact with the metal ion via a nitrogen atom or a sulfur atom and not via a carboxyl group [11]. However, to describe the metallization process of a peptide one needs affinities of the metal ion that correspond to the interactions of amino acids with the ion, not only through a carboxyl group (frequently this group is not accessible to the ion owing to the peptide bond formation), but also through other chemical groups present in the peptide side chains. These values may be obtained experimentally by determining affinities of the metal ion towards specific sites within the biomacromolecule or by studying C-protected and N-protected amino acids [12, 13]. A systematic experimental study of the zinc affinities towards all amino acids is, unfortunately, missing. An alternative to the experiment is to determine the affinity values using theoretical calculations such as those used in the present work.
In order to shed more light onto the mechanism of the metallization process and to identify possible metallization sites within macromolecules involving amino acids, we performed quantum chemical calculations of amino acid complexes with the zinc(II) ion. This ion plays an important role in various biochemical processes, and it is involved in numerous reactions in the areas of organometallic chemistry and materials science. An interesting feature of the zinc(II) ion is its ability to adopt a tetrahedral, a trigonal bipyramidal, or an octahedral geometry depending on the ligands bonded to the ion. While the Zn2+ aqua ion, as well as Zn2+ complexed by two N donors, is six-coordinated [14, 15], the zinc(II) ion coordinated by at least three N or S donors forms either tetrahedral or trigonal bipyramidal complexes [16].
Methods and models
All calculations in this work were performed using the density functional theory method with Becke’s three-parameter hybrid exchange functional and the Lee–Yang–Parr correlation functional (B3LYP) [17, 18]. This method was chosen because it combines a relatively low computational cost with a relatively high accuracy. A test in the extended G3 benchmark set (for molecules containing only first-row and second-row atoms) gives an average error of approximately 18 kJ mol−1 [19], while for transition metals the errors are believed to be in the range 12–20 kJ mol−1 [20].
Two different, standard basis sets were used: the medium-sized 6-31G* basis set and the large 6-311++G** basis set. In both cases a separate, full optimization of molecular systems was performed. Additionally, the energy was also evaluated for the 6-31G* optimized structures using the 6-311++G** basis set (denoted as 6-31G*/6-311++G**). The calculations were done for the systems immersed in water, whose presence was simulated using the polarizable continuum model approach [21]. For each amino acid molecular model a full optimization of all geometrical parameters was performed and the Hessian matrix was calculated in order to assess whether the predicted equilibrium geometry corresponds to a true energy minimum. Since it was not possible to converge the self-consistent-field calculations very tightly, small imaginary frequencies appeared in the calculations. They were considered insignificant and were ignored. The zero-point vibration energy corrections scaled by a factor of 0.96 were added to determine the final energy values [22]. The calculations were performed using the quantum chemistry package Gaussian 03 [23].
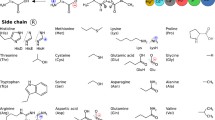
To save computational time we used simplified molecular models of amino acids that only include the most important fragments of the molecules from the viewpoint of the interaction with Zn2+. Such simplified models are commonly used in computational studies and, if the models are chosen correctly, the simplifications usually have little effect on the calculations [10, 24, 25]. In this study, we constructed models of all the amino acids that are expected to have high affinities towards the Zn2+ ion, i.e., arginine, asparagine, aspartate, cysteine, cystine, glutamine, glutamate, histidine, lysine, methionine, proline, selenocysteine, serine, threonine, tryptophan, and tyrosine (Fig. 1). For some of the amino acids two different models (a protonated model and an unprotonated model) were constructed (Fig. 1). Additionally, we performed calculations for a model of a peptide bond (both protonated and deprotonated).

Molecular models of amino acids and peptide fragments used in this work
To quantify the affinity of the metal ion towards amino acids, we considered the following model reaction (for all models except for aspartate/glutamate, cystine, and deprotonated peptide models)
or, if the ligand was a bidentate chelator (aspartate/glutamate, cystine, and deprotonated peptide), the reaction
where “aa” stands for the amino acid. The reactions in Eqs. 1 and 2 allowed us to determine the relative affinity of Zn2+ towards the amino acid with respect to the affinity of zinc(II) towards water. Identical reaction schemes and a similar computational approach were also used to determine the relative affinities corresponding to zinc(II) complexes formed with two or more amino acids.
Results and discussion
The results of the calculations for the zinc–amino acid interactions at the B3LYP/6-311++G** level of theory are summarized in Table 1 (the results from the two other approaches and the analysis of differences between them are available in the supplementary material). The highest affinity towards Zn(II) occurs for the deprotonated serine model (−96.8 kJ mol−1). Interestingly, there is no experimental evidence of an enzyme with a zinc ion complexed by at least one serine moiety in its active site. This phenomenon may be explained in terms of the deprotonation of serine; such a process is not likely to occur under physiological conditions owing to the large pK a value of the serine hydroxyl group (pK a = 13). Another reason may be a large affinity of the deprotonated serine towards Zn2+, which, at the enzymatic active site, may lead to a very rigid structure and low enzymatic activity. A similar argument may be applied in the case of the deprotonated peptide bond model (−66.7 kJ mol−1) and the deprotonated tyrosine (−39.7 kJ mol−1). The deprotonation of these systems may occur only at a very high pH. At such a pH the whole biosystem is likely to be completely inactive.
There are two other amino acid models with high affinities towards zinc(II): deprotonated cysteine (−60.4 kJ mol−1) and histidine (−34.3 kJ mol−1). The high affinity of cysteine and histidine should be of no surprise, since it was predicted earlier by experiment [26]. The experimental dissociation constant (at 25 °C in water) of the Zn–histidine complex is 8.76 × 10−13 [27], while the dissociation constant of the zinc–sulfide complex is 1.1 × 10−24 [28]. Assuming that the values of the dissociation constant are additive [10], we obtain the following affinity values in water: −34.4 kJ mol−1 for the Zn–histidine complex and −68.5 kJ mol−1 for the Zn–cysteine complex. The theoretical values of the zinc–amino acid affinities for those two amino acids predicted in the present work are in good agreement with these experimental data. These results also agree with the experimental data describing various enzymes which are known to bind zinc(II) via cysteine moieties [29].
Surprisingly, our calculations reveal that the affinity of the selenocysteine towards zinc(II) is small, even though the chemical properties of selenocysteine are similar to the properties of cysteine. Unfortunately, no experimental affinity data for the Se–Zn systems are available, since there is no evidence of an enzymatic active site with a selenocysteine residue.
As in the case of copper(II), zinc(II) has a high affinity towards the carboxylate group (a model of either glutamate or aspartate) [10]. Surprisingly, a moderately high affinity (−13.4 kJ mol−1) was also predicted for the proline residue complexing a zinc ion via the nitrogen atom of the five-membered ring. A similar affinity was predicted for the unprotonated lysine (−17.9 kJ mol−1), but this value is of little importance, since this amino acid residue does not exist in the unprotonated form under physiological pH.
Two more values need to be commented—the zinc affinity of the deprotonated selenocysteine and the asparagine/glutamine/peptide bond model interacting with a zinc ion via the oxygen atom. Surprisingly, our calculations reveal that the affinity of selenocysteine towards zinc(II) is small (0.23 kJ mol−1), even though the chemical properties of selenocysteine are similar to the properties of cysteine. Unfortunately, no experimental affinity data for the Se–Zn systems are available, since there is no evidence of an enzymatic active site with a selenocysteine residue. The value of the second affinity is even higher (9.63 kJ mol−1). On the other hand, our previous studies of copper(II) interactions with peptides revealed that interaction between asparagine/glutamine and metal ion may be of great importance in the metallization studies of macromolecules, where the metal ion binding relies on the interplay of two or more amino acid residues [10]. Therefore, it is important to note that the accuracy of the method used in this study is approximately 18 kJ mol−1 and more accurate studies are needed for those systems that fall within this range.
All other amino acid residue models (arginine, methionine, protonated histidine, protonated lysine, serine, trptophan, tyrosine) lack the ability to bind the zinc(II) ion owing to the positive value of the interaction energy and the inability to form a stable system with the ion.
Another interesting aspect of the Zn2+ interaction with the amino acids is the structure of the Zn2+–amino acid complexes. For most amino acid models studied in this work, the [Zn(H2O)5(amino acid)]2+ or [Zn(H2O)4(amino acid)]2+ systems remain in the octahedral geometry. This is, however, not the case for three amino acid residues. The deprotonated serine forces other ligands to adopt a tetrahedral geometry around the zinc(II) ion with only four ligands around the metal ion. The same is true for cysteine, which is in agreement with the known experimental crystal structures of some zinc-containing enzymes with the cysteine in the active site. In these enzymes, Zn2+ is always complexed by only four moieties that adopt a tetrahedral geometry around the ion. Here we should mention that the zinc complexes with the deprotonated selenocysteine and the deprotonated peptide bond are predicted to adopt a trigonal bipyramidal geometry.
The results of this part of the study allow us to suggest that the cysteine and histidine moieties have the highest affinity towards the zinc(II) ion under physiological conditions. On the basis of this result one can suggest where the process of the zinc metallization of a biomacromolecule would start just by looking at its three-dimensional structure and identifying the regions with a high density of the amino acid residues with high affinity towards the zinc(II) ion (cysteine, histidine, aspartate, glutamate, proline and also possibly selenocysteine, asparagine and glutamine). This information may be helpful in controlling the metallization process, as well as in designing nanotube-like peptides with high expected affinities towards zinc(II).
From the calculated interaction energies of systems with one or more histidine moieties (Table 2), it becomes clear why the [Zn(H2O)5His]2+ complex adopts an octahedral geometry. The relative interaction energy favors this complex over [Zn(H2O)3His]2+ by 13.9 kJ mol−1. Also, in the case of the [Zn(H2O)4(His)2]2+ complex, the most energetically favorable geometry is octahedral (by 24.6 kJ mol−1). On the other hand, the [Zn(H2O)3(His)3]2+ system favors the square-planar geometry by approximately 23 kJ mol−1 relative to the octahedral geometry. It is also interesting to note that, for this complex, there is a possibility of binding a fifth ligand (a water molecule or another amino acid moiety) using the site opposite the already bound water molecule. This result is in agreement with the experimental data, which suggest that for certain zinc enzymes a five-coordinated active site is a valid possibility. In this case, however, the Zn–O distance is relatively long (approximately 4.1 Å), suggesting that the second water molecule is only weakly bound. The visualization of the optimized geometry of the system suggests that the three imidazole moieties already create such a steric hindrance that the space for yet another molecule is significantly reduced (Fig. 2).

Optimized geometry of the [Zn(H2O)3(imidazole)3]2+ system
Conclusions
The results of this work contribute to the understanding of the metal ion–biomacromolecule interactions. The work has concerned the affinities of Zn2+ towards all amino acid residues and the structural features of the complexes formed owing to the Zn2+–amino acid interactions. The computational approach used in this work may become a useful tool for predicting the metallization sites for other metals and other biomolecules. A relatively fast, yet fairly accurate computational approach, such as the one used in this work, may guide experimental efforts concerning the metallization of biomacromolecules by predicting possible metallization sites and their affinities towards different metal ions.
The results of this investigation suggest that serine-rich and cysteine-rich sites exposed to the solution are the most likely targets for the first phase of the metallization process by zinc. Other sites, however, cannot be ruled out, since several other amino acid residues also possess substantial affinities towards zinc(II). In this work all moieties present in a typical protein system and possessing positive affinity towards Zn2+ have been identified.
Additionally, the coordination geometry around the Zn2+ ion in typical enzymatic active sites was investigated. It was found that even a single cysteine moiety forces the zinc(II) coordination system to adopt a tetrahedral geometry. On the other hand, zinc complexes with one or two histidines adopt an octahedral geometry, but they prefer a tetrahedral geometry when the number of histidines becomes greater than two. This result may be helpful in biochemical studies of active sites of zinc-containing enzymes and possibly also for developing new active compounds interacting with those sites.
References
Mitchell P (1976) Biol Rev 41:445–502
Blankeship RE (ed) (2002) Molecular mechanisms of photosynthesis. Blackwell, Oxford
Berridge MJ, Bootman MD, Lipp P (1998) Nature 395:645–648
Tajmir-Riahi HA, Langlais M, Savoie R (1988) Nucleic Acids Res 16:751–762
O’Halloran TV (1993) Science 261:715–725
Orvig C, Abrams MJ (1999) Chem Rev 99:2201–2204
Jurisson SS, Berning D, Jia W, Ma D (1993) Chem Rev 93:1137–1156
Braun E, Eichen Y, Sivan U, Ben-Yoseph G (1998) Nature 391:775–778
Yang Y, Constance BH, Deymier PA, Hoying J, Raghavan S, Zelinski BJJ (2004) J Mater Sci 39:1927–1933
Trzaskowski B, Deymier PA, Adamowicz L (2006) J Mater Chem 16:4649–4656
Hallman PS, Perrin DD, Watt AE (1971) Biochem J 121N:549–555
Michael SF, Kilfoil VJ, Schmidt MH, Amann BT, Berg JM (1992) Proc Natl Acad Sci USA 89:4796–4800
Corradi AB (1992) Coord Chem Rev 117:45–98
Munoz-Páez A, Pappalardo RR, Marcos ES (1995) J Am Chem Soc 117:11710–11720
King RB (ed) (2006) Encyclopedia of inorganic chemistry, 2nd edn. Wiley, New York
Zhang C, Janiak CJ (2004) Chem Crystallogr 31:29–35
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Blomberg MRA, Siegbahn PEM, Babcock GT (1998) J Am Chem Soc 120:8812–8824
Curtiss LA, Raghavachari K, Redfern RC, Pople JA (2000) J Chem Phys 112:7374–7383
Miertus S, Scrocco E, Tomasi J (1981) Chem Phys 55:117–129
Johnson RD III (ed) (2005) NIST computational chemistry comparison and benchmark database, NIST standard reference database number 101, release 12
Frisch MJ et al (2003) Gaussian 03, revision B.03. Gaussian, Pittsburgh
Siegbahn PEM, Blomberg MRA (1999) Annu Rev Phys Chem 50:221–249
Siegbahn PEM (2001) Theor Chem Acc 105:197–206
Myari A, Malandrinos G, Deligiannakis Y, Plakatouras JC, Hadjiliadis N, Nagy Z, Sovágó IJ (2001) Inorg Biochem 85:253–261
Chen W, Sammynaiken R, Huang YN (2000) J Appl Phys 88:5188–5193
Yi G, Sun B, Yang F, Chen DJ (2001) Mater Chem 11:2928–2929
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) Nucleic Acids Res 28:235–242
Acknowledgements
NSF grant no. 0303863 and CPU time from University of Arizona supercomputing center are gratefully acknowledged. Figure 2 was generated using the xyzviewer software designed by Sven de Marothy.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Trzaskowski, B., Adamowicz, L. & Deymier, P.A. A theoretical study of zinc(II) interactions with amino acid models and peptide fragments. J Biol Inorg Chem 13, 133–137 (2008). https://doi.org/10.1007/s00775-007-0306-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-007-0306-y