
Abstract
After a review of the current status of density functional theory (DFT) for spin-polarized and spin-coupled systems, we focus on the resting states and intermediates of redox-active metalloenzymes and electron transfer proteins, showing how comparisons of DFT-calculated spectroscopic parameters with experiment and evaluation of related energies and geometries provide important information. The topics we examine include (1) models for the active-site structure of methane monooxygenase intermediate Q and ribonucleotide reductase intermediate X; (2) the coupling of electron transfer to proton transfer in manganese superoxide dismutase, with implications for reaction kinetics; (3) redox, pK a, and electronic structure issues in the Rieske iron–sulfur protein, including their connection to coupled electron/proton transfer, and an analysis of how partial electron delocalization strongly alters the electron paramagnetic resonance spectrum; (4) the connection between protein-induced structural distortion and the electronic structure of oxidized high-potential 4Fe4S proteins with implications for cluster reactivity; (5) an analysis of cluster assembly and central-atom insertion into the FeMo cofactor center of nitrogenase based on DFT structural and redox potential calculations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The great versatility of transition metal centers in biological systems is well known [1]. Many of the reactions of transition metal complexes at the active centers of enzymes are controlled by the chemistry of their high-spin metal ions and bonded ligands [2]. The comparatively greater prevalence of high-spin metal sites is connected to the dominance of first-row transition metal ions, and to the type of ligands bound. In contrast to organometallic chemistry, carbon coordination is comparatively rare. Ligands can be amino acid side chains (N, O, or S bonded to the metal), main chain peptide groups, cofactors, and/or simple groups like sulfide, oxide, hydroxide, or substrate molecules. Second-shell, third-shell, and more extended group interactions can also be very important, particularly when the active site is charged. This is shown, for example, by mutagenesis and kinetics studies in Mn, Fe, and CuZn superoxide dismutases (SODs) [3–6]. Highly charged active sites are not that unusual, particularly when these sites can be strongly stabilized by main chain or side chain hydrogen bonds. The premier examples are the many dinuclear or polynuclear iron–sulfur clusters in FeS proteins [7]. These clusters are usually multiply charged anions that can be either deeply buried in the protein interior or near (but not on) the protein surface depending on the system [8]. From a theoretical/computational standpoint, these long-range charged or polar interactions are difficult to treat with proper accuracy, but major progress has been made on this problem by combined quantum mechanical and electrostatic methods. There are related efforts with combined quantum mechanics (QM)/molecular mechanics (MM) methods, and advances utilizing molecular dynamics. For a more detailed account of methods for treating the longer-range protein and solvent environment interacting with the quantum active-site metal complex, see [2].
Correspondingly, the viewpoint put forward by some structural biologists that proteins have a predominantly hydrophobic interior and a hydrophilic exterior is demonstrably a vast oversimplification for both metalloenzymes (FeS clusters, FeO dimer enzymes, SODs) and non-metal-containing enzymes (protein tyrosine phosphatases) [9, 10]. In a number of cases, charged substrates gain access to interior metal sites via water channels (wide or narrow), mobile loops, or flaps controlling access to the active-site pocket. Further, the view that proteins can be designed on the basis of overall shape alone (folding analysis) is severely limited, as is clear from the metalloprotein design work of Hellinga and others [11, 12]. The design must reflect the metal–ligand interactions with high priority, so that an inside-out design is required although this may be based on well-defined and known protein scaffolds. As with natural enzymes, redesigned enzymes must avoid the pitfalls of maladaptive functions. However, exploratory mutagenesis or the use of alternative substrates can often reveal unexpected functional capabilities [13], and allow for a broader understanding of the multiple requirements (sometimes conflicting) that the active site and entire enzyme must fulfill. Quantitative evaluation of interaction energies among protein fragments and the active-site cofactor is closely linked with analysis of protein function and design whether in native, mutated, or entirely synthesized structures. Such analysis provides a testing ground for theoretical/computational methods, allows for practical applications of experimental and theoretical methods, and leads to generalizations about principles governing protein fragment and cofactor interactions. In all of this, solvation often plays an essential role, involving either discrete bonded waters, waters filling small cavities, or bulk water. Solvation effects are one major source of nonadditivity in cofactor–protein and protein internal interactions. (The complex shape of the solvent region produces energies of interaction of the solvent and protein with the active-site metal complex that are not decomposable into a simple sum of terms. In the framework of the linear Poisson–Boltzmann equation, the “protein field term” is decomposable, but the “reaction field term” is not, and even the protein field term includes solvent screening effects [14, 15].) Other major sources of complexity include proton-coupled electron transfer [5, 16–20] and cooperative charge-coupled proton transfer [9, 10]. These may involve motions of amino acid side chains [21] as well as substrates and larger-scale domain motions in some cases. (See Nicholls and Ferguson’s [22 discussion of respiratory chains and especially of the motion of the Rieske iron–sulfur protein within the cytochrome bc 1 complex.)
For example, iron–sulfur proteins often bear a large negative protein surface charge from surface carboxylates (unrelated to the active-site iron–sulfur cluster charge) [7, 23]. Solvent screening strongly reduces the influence of the surface charge on the cluster redox potential compared with second-shell hydrogen bonding, and general dielectric (protein plus solvent) response (orientational and electronic polarizability) [14]. At the same time, such a large negative surface charge may facilitate protein–protein (or protein–coenzyme) recognition for electron or proton transfer and such docking may modulate the redox potential for electron transfer [23]. Further, the interaction energy between the active-site iron–sulfur cluster anion and the protein–solvent environment is very large, so a proper accounting of this is needed for predicting redox potentials [14].
Many metalloenzymes are redox-active with one or multiple electron transfers intrinsically associated with the catalytic transformation and these can have coupled proton transfers as well. In some cases, first- or second-shell ligands (or more distant groups) can be “redox noninnocent” during or as a result of the catalytic or activation cycle; for example, porphyrin, tyrosine, tryptophan, cysteine, and glycine radicals have been observed, as well as the more commonly known coenzyme species like semiquinones or substrate radicals like superoxide (O2 −) or nitric oxide (NO). Chemistry of a low-spin type is much less prevalent, but it does occur at the catalytic center of hydrogenase [24], in nitrile hydratase [25], and some other enzymes [vitamin B12 (cobalt)] [26]. In other enzymes, both high-spin states and low-spin states can be involved at different stages in the catalytic cycle, as in cytochrome P450s [27] and related heme enzymes, or in carrier proteins like hemoglobin, and myoglobin [28, 29]. Redox changes can also be the trigger for substantial spin-state changes and so can ligand binding. Where high-spin metal sites are involved, there may be one, two, or more metal sites, which allows for possible spin coupling between sites owing to the bridging ligands. Complexes of this type include the active sites of polynuclear iron–sulfur proteins [7], the water-oxidizing complex of photosystem II [30], iron-oxo dimers including methane monooxygenase (MMOH) [31], ribonucleotide reductase (RNR) [32], and hemerythrin [33], and even much larger assemblies like ferritin [34].
In this commentary, our focus will be on resting states and intermediates of redox-active metalloproteins—their structures, protonation, and redox states, physical properties, and energies. Transition states link intermediates and both are essential for kinetics. However, if an incorrect intermediate is identified, the transition state linking this to another state may well prove irrelevant. In most proteins, whether enzymes, regulatory proteins, or structural proteins, X-ray structures are only available for a few states of a catalytic or regulatory cycle. Spectroscopic studies augment the information available from X-ray structures, and provide vital data for characterizing intermediates and transition states (also using kinetics methods). The analysis and interpretation of spectroscopies is often not direct, and there may be real or apparent contradictions between different spectroscopic tools. High-quality QM methods are very useful for structure, energy, and spectroscopic properties evaluation, so that a coherent analysis of physical state properties can be obtained. The methods employed need to be adequate for large systems, which brings us to density functional theory (DFT). Further, construction of appropriate model systems for QM calculations involves a number of subtle issues. In particular, where the structures of intermediates are not known, QM model structures (including also the surrounding environment) are proposed as “computational hypotheses” to be tested against experimental data. Such multimodal analysis can provide tests of DFT, lead to new ideas and experiments, organize disparate phenomena into a conceptual framework, or point out experimental problems. The systems examined include transition metal complexes as well as metalloproteins, since often more complete information is available for metal complexes than for proteins. Metal complexes can also be synthesized that exhibit wider diversity in properties and energetics. In tests against “simple” metal complexes, the quality of DFT predictions can be critically examined. Further, a systematic overestimation or underestimation of energies is easier to account for when this is well characterized for a “test” set of known oxidation states, structures, and spin states.
Density functional theory overview
To calculate and critically analyze these states and reaction pathways, flexible and accurate electronic structure methods are needed. The difficulties stem from a number of linked problems. Most fundamentally, the first-row transition metal ions have a very compact 3d electron shell, which produces strong electron–electron interactions, and requires a good treatment of electron correlation, balanced with the treatment of electron exchange. Metal–ligand bonding can be strong or weak field, with resulting effects on spin states, and metal–ligand covalency. This problem is worse here than for the second or third transition metal series. (Conversely, relativistic effects are smaller for the lighter first-row metal series.) In practice, standard Hartree–Fock (HF) methods are totally inadequate for this task since electron correlation is neglected, and DFT methods have taken over the primary role. These are parameterized from atomic and, in some cases, from simple molecular energies to give a good account of electron exchange combined with correlation effects. The functional forms used are derived from a number of physical limits and principles, and many different exchange–correlation potentials and energy expressions have been generated. For the present, we will classify these into the following families [35]: (1) local spin density approximation (LSDA) exchange–correlation potentials; (2) generalized gradient approximation (GGA) corrected exchange–correlation potentials;Footnote 1 (3) hybrid potentials, which involve an explicit mixture of a percentage of HF exchange combined with a complementary percentage of GGA exchange plus correlation; (4) meta-GGA or hybrid-meta-GGA, which includes an explicit electronic kinetic energy contribution to exchange and correlation along with HF and GGA terms.
Exchange and correlation potentials
The movements of electrons in molecules are accompanied by instantaneous changes in the electronic distribution. The fundamental behavior of these conditional probability densities is governed the theory of the second-order reduced density matrix [36]. Compared with the average electron density, ρ, there is a deviation called the “hole density,” which is carved out in the vicinity of the reference electron [37]. There are two types of hole functions: (1) the exchange (or Fermi) hole and (2) the opposite spin correlation (or Coulomb) hole. A more precise mathematical account of these issues is given in recent reviews [38, 39]. The Fermi hole has the largest influence and will be examined first.
The exchange hole (or Fermi hole) creates a hole of the same spin as the reference electron (an α hole in the α electron density for a reference electron of spin α, and a β hole for spin β). For any reference electron, the Fermi hole function integrates to one positively charged electron (or −e −, where e − is the electron charge); this density is maximum at the reference electron, and dies off largely monotonically with distance. The Fermi hole in HF theory differs from those in DFT in two ways: (1) the HF Fermi hole is nonlocal—it is different for every molecular orbital; by contrast, in pure DFT, the Fermi hole is the same for all α electrons, and there is a different Fermi hole which is the same for all β electrons; (2) the HF hole functions are more diffuse than in DFT-LSDA or GGA theories for molecules [40]; because the LSDA or GGA exchange hole functions are more confined (more localized), there is an associated energy lowering particularly for moderate to weak covalent bonds. This energy lowering typically gives improved bond energies for homolytic cleavage of an electron pair bond, and it is said that LSDA or GGA DFT exchange contains “some left–right correlation” (also called static correlation) in contrast to HF. A GGA-type exchange potential is a function both of the local electron density for a given spin, and of the gradient of electron density of that spin at the position of the reference electron (with the same spin). It is typical that molecules calculated at the HF level exhibit both underbinding and too-short bond lengths compared with experiment, as well as too-high vibrational frequencies, while LSDA molecules show overbinding and too-short bond lengths. GGA-described molecules give improved bond energies (still somewhat overbinding) and typically very good geometries. At a purely empirical level, it is not too surprising that hybrid potentials combining HF and DFT-GGA have often been very successful for bonding and reactions of main group molecules [41]. The justification for hybrid potentials like B3LYP, however, goes further by including a correlation energy contribution to the electronic kinetic energy via the “adiabatic connection theorem” [37]. B3LYP yields an excellent description of main group thermochemistry, and often reasonable results for transition states. Transition metal complexes, however, are much more complex electronically than most main group molecules, and many problems related to energies and reaction pathways are only partly understood.
The Coulomb hole is a hole function of opposite spin to that of the reference electron (an α spin reference electron produces a hole in the β spin electron density). The Coulomb hole function integrates to zero positive electrons, because the positive density induced very near the reference electron position (called the “electron cusp”) is compensated by enhanced negative hole density farther away. Density functional correlation potentials are supposed to represent this highly local correlation of opposite-spin electrons, called “dynamic correlation” near the cusp of the reference electron, but not longer-range “static” or “left–right” correlation, which also involves opposite-spin electrons. It is now clear, however, that the performance of exchange–correlation potentials depends on the combined behavior of the exchange and correlation parts of the potential and that deficiencies in one part may be (partly) compensated by the type or mixing coefficient of the alternate part [41]. The convention for naming exchange–correlation potentials is an alphabet soup, with the exchange potential named first (by a one- or few-letter and number acronym) and the correlation potential named second (similarly).
The most important gradient-based exchange potentials derive from the work of Becke (1986, 1988) (called B86, B88), and depend on both the local spin-dependent density ρ σ , where σ = α or β (spins), and on x σ = |∇ρ σ |/ρ σ 4/3. Both the B88 and the later Perdew–Wang 1991 (PW91) exchange potentials have rather similar form and are similar for small x σ , but deviate for larger x σ . Two other related exchange energy expressions (and potentials) are PBE [42], which is a simplification of PW91, and Gill 1996 (G96), which is Gill’s simpler alternative to B88 [43].
The gradient-dependent potential adds to the local exchange potential of Slater and Dirac [44], proportional to ρ α 1/3 for α spin electrons and ρ β 1/3 for β spin electrons. For Slater exchange, the total exchange energy takes the form −C x ∫ρ α 4/3 − C x ∫ρ β 4/3. In the LSDA, appropriate for an electron gas, C x (LSDA) = (3/4)(6/π)1/3, but Slater exchange is more general C x (Slater α) = (3α/2)C x (LSDA), where α is a parameter between 2/3 (for heavy atoms) and 0.77, 0.78 (for He and spin-polarized H, respectively). In much of the early work using the Amsterdam Density Functional (ADF) code and its precursor, a typical average value was used for all atoms, α = 0.7, which gives 1.05C x (LSDA) [45]. The reason for bringing up this ancient history is the close connection to an improved GGA potential of Handy and Cohen [40], which is similar in spirit and results to a number of recent comparable attempts to improve both atomic energies and molecular energetics.
In recent and important work, Cohen and Handy [40] proposed a new DFT exchange energy form by moderately generalizing the B86 exchange term and reweighting the linear combination of LSDA exchange energy with gradient-term exchange energy, to fit atomic HF energies better than either LSDA plus B88 or (LSDA plus B86). The resulting potential also improves bond dissociation energies for small main group molecules particularly when bonds are stretched (1.25 or 1.5 times their equilibrium distance) with no further adjustment. This confirms that GGA potentials like B88 and the Handy–Cohen potential (called OPTX or O) have left–right correlation energy; comparison with multiconfigurational self-consistent-field (SCF) calculations for left–right correlation energy in six small molecules, and evaluation of atomic energies, molecular binding energies, and geometries for a test set of 93 atoms and molecules (main group chemistry) show that OPTX combined with dynamic correlation potentials (LYP or PW91 → OLYP or OPW91) gives improved results compared with BLYP or BPW91, of comparable quality to B3LYP.
Xu and Goddard [46] have combined the exchange functionals from B88 and PW91 into a new functional (called X), with large x σ behavior between those of B88 and PW91, which leads to considerably improved atomic and molecular energies for the LYP correlation potential, particularly for the hybrid X3LYP potential. O3LYP is of similar quality and both (also OLYP) are preferable to B3LYP for van der Waals interactions, and are of similar quality for thermochemical energies. For small main group molecules, ionization potentials and electron affinities are of good and comparable quality, 0.1–0.2 eV for BLYP, OLYP, B3LYP, and O3LYP, and predicted proton affinities are also good, with mean errors of 1.4 kcal mol−1 for OLYP and B3LYP, 1.1 kcal mol−1 for O3LYP, and 1.9 kcal mol−1 for BLYP. X3LYP performs better than B3LYP and O3LYP for a hydrogen-bonding test case (H2O dimer), and X3LYP and XLYP perform comparably well to B3LYP for s → d promotion energies for first row-transition atoms and (1+) cations.
Recent work on small metal–ligand complexes and metal–metal dimers
Schultz et al. [35, 41] have recently studied the bond lengths and bond energies for a relevant group of small transition metal complexes and bare metal dimers (M2 or MM′) using a wide variety of density functionals (LSDA, GGA, hybrid, and meta-hybrid). A number of these metal complexes are spin-polarized, and some of the transition metal diatomics are ferromagnetically or antiferromagnetically coupled. Atomic ionization energies for five first-row metal atoms and C, O were also included. In these groups, they also identify systems that are intrinsically multiconfigurational (having a number of nearly degenerate or low-lying configurations). These systems will have large static correlation energies, and will differ strongly from the HF approximation in both geometry and bond energies. These include transition metal diatomics (Cr2, Ni2, V2, and Mo2), but also molecules (FeS, FeO, CoOH+, MgO, and VO). Such bonding is relevant to FeS-, FeO-, and Co- containing metalloproteins. Particularly because of these multiconfigurational cases, the overall “best performing functionals” for energies and bond lengths (using a weighted average) are mainly GGAs (five out of six), including G96LYP, XLYP, mPWLYP, and BLYP, and some functionals of their own construction MOHLYP and hybrid MPWLYP1M. The mean energy errors (kilocalories per mole) for some relevant potentials are as follows: 4.4 (OLYP), 5.7 (BLYP), 6.0 (XLYP), 6.4 (BPW91), and 8.4 (BP86) for GGAs, and 5.1 (MPWLYP1M), 6.2 (O3LYP), 8.1 (X3LYP), and 8.3 (B3LYP) for hybrids. The best performing hybrid (MPWLYP1M) has only 5% HF exchange, and functionals like B3LYP (20%) and X3LYP (22%) give larger energy errors because of HF mixing.
Two cautionary notes are relevant in assessing these results. First, these transition metal diatomics were calculated with the broken-symmetry (BS) method where needed (for Cr2 and Mo2), but spin projection to the proper “pure spin” ground state was neglected. This makes a major difference, and the effect is much different for hybrid methods versus GGA. Spin projection was shown by Edgecombe and Becke [47] to have a major effect in improving the binding energy, the geometric minimum, and the entire potential energy curve for Cr2 using a hybrid B3P86 potential, while the BLYP binding energy gets worse on going from the BS solution to the spin-projected result. The Mo2 case will probably also be improved with BS plus spin projection for hybrid potentials, while the GGA potentials do not break symmetry at or near the equilibrium geometry. This neglect of spin-projection effects will skew the performance of hybrid versus GGA potentials for these transition metal diatomics (two out of nine molecules). Second, one can ask how representative small transition metal–ligand molecules like FeS and FeO are compared with the much higher coordination number transition metal complexes observed in biological systems or with transition metal complexes in solution. Very small transition metal complexes (like diatomics) often have high degeneracies or near degeneracies; these degeneracies are split significantly in higher coordination, and further split in low-symmetry systems (typical of biological systems). Higher-coordination metal complexes will then have less “multiconfigurational character.”
Left–right correlation: its contents and discontents
In the Cohen–Handy paper [40] mentioned earlier, the advantages of approximate inclusion of left–right correlation implicit in local DFT exchange functionals were analyzed. This type of work has a long history, with prominent contributions by Gritsenko and Baerends [48, 49], and earlier by Tschinke and Ziegler [50], going back to Slater [51]. Our group has also contributed to this problem for metal–metal bonding interactions [52] and core–hole localization versus delocalization [53]. While left–right correlation is implicitly present in LDA- and GGA-type exchange potentials, there are both advantages and difficulties here. Such left–right correlation represents a moderate to weak electron pair bond, but as the bond weakens further, symmetry breaking occurs; then the left–right correlation is represented explicitly both in the spatial and spin wavefunction and in the corresponding energy. Further, BS allows for the buildup of spin polarization from different electron orbitals on each metal center, which cannot be captured by the implicit left–right correlation in the exchange potential. BS combined with implicit left–right correlation should lead to some double counting of the bonding interaction associated with spin coupling, and calculations of Heisenberg J parameters typically give too-strong antiferromagnetic coupling compared with experiment, as we will discuss later.
A further problem is that left–right correlation occurs between electrons of opposite spin, while an exchange (Fermi) hole represents the interaction of electrons of the same spin. This is not a problem where the α spin electron density is the same as the β spin electron density (in a “spin-restricted” framework), but it is a problem when spin polarization is allowed (and spin polarization is often important). This contributes to the problem of finding properly balanced exchange–correlation potentials for spin-polarized and for spin-coupled systems.
As a final comment on this topic, we should mention that not all systems are describable solely in terms of pair bonds and lone pairs. In transition states, in radical reactions, and in spectroscopies generating odd electron species, the implicit left–right correlation can cause problems. As Gritsenko and Baerends point out, energy difficulties arise when the number of electrons (n) involved in a bond is not divisible by the number of centers involved (more precisely, by the number of relevant fragment orbitals, m). Some symmetric SN2 reactions fit this category since there are three-center four-electron bonds in the transition state (n = 4, m = 3, n/m = 4/3), and related problems occur for two-center three-electron bonds (n = 3, m = 2, n/m = 3/2) as seen for some cation and anion radicals [48], for core holes in symmetric molecules (like N2 +) [53], and for valence lone-pair holes in symmetric molecules resulting from ionization [54]. In these cases, spatial symmetry breaking provides a partial solution for obtaining better energies. These problems arise more frequently in main group chemistry and spectroscopy than in transition metal complexes, because the latter have higher electron densities, are typically less symmetric, and often exhibit both spin and space symmetry breaking.
Spin-polarized complexes
As mentioned before, many biologically relevant transition metal complexes and transition metal ions in solution are high spin. There are large spin-polarization effects, and the majority-spin potential on the metal ion will differ dramatically from the minority-spin potential, with smaller but significant effects propagating to the ligands. DFT studies have shown that high-spin Fe, particularly Fe(III), can lead to an inverted-level scheme with filled ligand levels sandwiched between lower-lying majority-spin metal levels (filled) and higher-lying minority-spin metal levels [empty for Fe(III) and mainly empty with a few filled levels for Fe(II)]. This condition exists for many mixed-valence dinuclear and polynuclear systems (iron–sulfur clusters), and is strongly supported by spectroscopic studies [39, 55]. For iron-oxo and manganese-oxo complexes, the level scheme is highly mixed, with ligand levels highly interspersed with minority-spin metal levels [56]. In solid state terms, these are narrow bandgap semiconductors bordering on semimetallic behavior.
While a number of standard DFT methods of GGA or hybrid form perform well in describing metal–ligand bonding for a given metal ion spin state (high spin, low spin, or intermediate spin), the electronic structure of the complex can change substantially with a change in spin state. Recently, the relative energies of different spin states (spin-crossover issues) have come under intense scrutiny, with a focus on improving the power of GGA, hybrid, and meta-GGA methods for predicting the correct ground-state spin. We will review this topic, and also discuss some alternative strategies even where calculated spin-crossover energetics are not reliable.
Spin-crossover problems
Swart et al. [57] have studied the relative spin-state energies of seven Fe complexes [three Fe(III) and four Fe(II)] using a wide variety of exchange–correlation potentials, including LSDA, GGA, hybrid potentials, and meta-GGA potentials. In particular, the OPTX exchange potential was examined in combination with LYP and PBE for correlation (OLYP and OPBE). These are five- or six-coordinate systems with mainly S, N ligands, but also some additional C, P, or Cl ligands. All the spin ground states of these complexes have been experimentally determined. All DFT functionals give the correct spin ground state in six out of seven cases, but the predicted spin-state energy spacings differ. The GGA-type OPBE gives the correct spin ground state for all systems, including the most difficult case, an Fe(III) complex, and displays spin-state energy spacings similar to those for B3LYP and other hybrid and meta-GGAs. Deeth and Fey [58] have performed a related study on different Fe(II) and Fe(III) complexes using a different assortment of GGA exchange–correlation potentials. The main outstanding problem here is that while the spin ground states have been experimentally determined, much less is known experimentally about the energy spacings of the different spin multiplets since these are almost always larger than accessible thermal energies and it is difficult to find proper spectroscopies to obtain relative energies of S = 1/2, 3/2, and 5/2 for Fe(III) complexes and S = 0, 1, and 2 for Fe(II) complexes.
Groenhof et al. [59] applied similar methods to examining the spin multiplet states of Fe(II) porphyrins, Fe(III) porphyrins, and Fe(III) porphyrin complexes with additional axial (SH)−, OH−, and H2O ligands. Again the spin-state spacings shown by OPBE are similar, although not identical to those of B3LYP, and the OPBE spin ground-state multiplet agrees with experiment where this is known. For both Fe(II) and Fe(III) porphyrins, the spin-state ordering is intermediate spin < high spin < low spin. Addition of an (RS)− ligand makes the high-spin state the ground state with either OPBE or B3LYP for Fe(III), and this persists on one-electron reduction to Fe(II). These results have significant implications for the catalytic cycle of cytochrome P450 enzymes, and further work is ongoing with QM/MM methods.
Spin-coupled complexes
There are many ligand-bridged dinuclear and polynuclear complexes of biological relevance. Metal–ligand radical complexes may also be spin-coupled. An antiferromagnetically coupled transition metal complex within DFT is usually represented by a BS state, where the spin-up (α) electron density occupies a different region of space from the spin-down (β) electron density. In general, there is overlap between these densities. The integrated spin density may sum to zero (when the ground state is a singlet S t = 0), or may be nonzero either owing to different site spins or to some non-anti-ferromagnetic alignments. In some cases, the totally spin aligned high-spin state may be the ground state, and the BS state lies higher in energy.
To be concrete, consider a system with two coupled site spins (either a dinuclear transition metal complex or a transition metal–radical complex). Then for site spins S 1 and S 2, QM allows for all possible total spin states ranging from S min = |S 1 − S 2| to S max = S 1 + S 2 in integer steps. This expression is called “the triangle inequality.” Ordinarily the pure spin states form a Heisenberg ladder obeying the Lande interval E(S) − E(S − 1) = JS assuming a Heisenberg Hamiltonian of the form H spin = J S 1 · S 2 monotonic with the total spin S, whether J is antiferromagnetic, J > 0, or ferromagnetic, J < 0. (Beware: there are various conventions for H spin and J, including H spin = −2J S 1 · S 2 , common for iron-oxo and manganese-oxo dimer complexes, and H spin = −J S 1 · S 2 , while the convention H spin = J S 1 · S 2 is common for iron–sulfur systems.)
More complicated cases are well known and important. They stem from three or more causes (or combinations thereof):
-
1.
There may be multiple metal sites with either the same or different J couplings. Then it may be energetically advantageous not to fully align or oppositely align all spins. The result is an intermediate spin state either for the total complex or for some set of subspins (like pair spins S ij ). This phenomena is called “spin frustration” and the resulting ground state is said to be “spin canted” with respect to either the total spin or subspins.
-
2.
For mixed-valence complexes, parallel alignment of spins (ferromagnetic alignment) is associated with enhanced electron delocalization and associated energetic stabilization for states with higher pair spins. When this is fully operative, a total high-spin alignment for the relevant pair occurs. However, often the intrinsic Heisenberg interaction is antiferromagnetic and vibronic trapping or solvent effects may also favor a “partially localized” state. An intermediate (or low) pair spin ground state may then result from this compromise.
-
3.
For a pair, one or both site spins may be less than their maximum. This can even happen when the metal ion sites are equivalent or nearly equivalent. We call this phenomena a “resonance spin crossover pair” (RSCP). It is physically distinct from “spin canting” even though the pair spin can be the same.
Compared with the complete Heisenberg ladder of spin states, the BS state is simpler, and represents a weighted average of spin states, emphasizing those of lower total spin. For dominant antiferromagnetic coupling, the BS state is “spin-bonding,” while the spin-aligned high-spin state is “spin-antibonding.” For simplicity, consider a transition metal dimer with equal site spins (S 1), and total spins ranging from S min = 0 to S max = 2S 1. In this dimer, the energy difference between high spin and the singlet ground state is
By contrast, the energy difference between high spin and BS is
A more general equation for either unequal or equal site spins is
Within DFT, the left-hand side is directly computed and used to obtain the J parameter. Meanwhile the corresponding pure spin states have relative energies
and the entire Heisenberg ladder can be constructed. Correspondingly,
Similar equations can be used for polynuclear complexes with multiple J parameters. In addition to the DFT spin-coupling evaluation being feasible, a comparison of E(HS) versus E(BS) with states derived from the triangle inequality shows the conceptual and practical simplicity of the E(BS) “spin interpolation” method compared with direct evaluation of all spin states E(S). For polynuclear systems, where there are a number of possible BS states, there are a vastly larger number of pure spin states, since the triangle inequality must be applied to one or more pairs of site spins, then triples, etc., up to the total system spin. (There are many ways to do this, but all yield these very large numbers of spin states.) By contrast, with the “best” (lowest-energy or “best-properties”) BS states as a guide, the “best” pure spin-state energies can be evaluated as discussed before, and properties can be calculated using spin projection operator methods based on the Wigner–Eckart theorem for vector operators. Only a small subset of spin states needs to be analyzed.
We have so far neglected the delocalization energy for a delocalized mixed-valence pair (of total pair spin S) given by
This result, called “double exchange” (later named “resonance delocalization coupling,” or “spin-dependent electron delocalization”), was originally discovered by Anderson and Hasegawa [60, 61] based on Zener’s earlier qualitative work, and then rediscovered by us (Noodleman and Baerends) [62] in the context of dinuclear and polynuclear iron–sulfur complexes using a simpler form of spin algebra (simple Clebsch–Gordon algebra instead of Racah algebra). Also, Girerd et al. [63, 64, 65] recognized the relevance of Anderson and Hasegawa’s work. Further, the pairwise delocalization in 4Fe4S systems was qualitatively anticipated by Middleton et al. [66] from experimental work on proteins, by Holm’s group [67] for synthetic analogues, and by Aizman and Case [68], on the basis of early DFT calculations. The resonance energy splitting increases with increasing total spin, and can be directly computed for the high-spin S = S max state within DFT to yield the resonance B parameter. If delocalization is suppressed (partially quenched) by other energy terms, including vibronic trapping, site inequivalence energy, or solvation energy, a simple matrix expression or perturbation theory can be used to derive the partial delocalization energies and corresponding eigenstates [64, 65, 69]. A surprising example of this will be analyzed later. Usually, pairwise delocalization is dominant for tetranuclear (4Fe4S) complexes, while trapping or partial delocalization occurs often for 2Fe2S complexes. For iron-oxo dimers and for manganese-oxo dimers or tetramers, site asymmetries and vibronic forces are typically dominant. We will consider a number of cases later on.
Structures of transition metal complexes
The predicted structures of transition metal complexes are usually quite good using density functional methods. In earlier work [70], we optimized the geometries for a series of simple mononuclear complexes including high-spin M(H2O)6 n+, low-spin M(CN)6 n−, and high-spin M(SCH3)4 n−, where the transition metals, M, are Mn2+,3+,Fe2+,3+, and Cu+,2+. The results were compared with experimental metal–ligand bond lengths for these 16 complexes. The exchange–correlation potentials used were LSDA (specifically Vosko–Wilk–Nusair, VWN, potential), a local potential that eliminates parallel spin correlation in the VWN potential (called VWN–Stoll, leaving only local Slater exchange and VWN opposite spin correlation), and the more elaborate (but now quite old and established) GGA potential, B88–Perdew 1986 (BP), which modifies the local parallel spin exchange interaction (Slater exchange) and modifies opposite spin correlation. The experimental metal–ligand bond lengths range from 1.9 to 2.5 Å. All three potentials predict geometries well, with average absolute errors ranging from 0.04 to 0.06 Å. As expected, VWN bond lengths are too short, consistent with overbinding by this local potential, while BP bond lengths are too long, and VWN–Stoll bond lengths lie in-between (overall too short).
We also examined 11 ligand-bridged dinuclear transition metal complexes containing manganese-oxo, iron-oxo, copper-oxo (and copper-peroxo), and iron–sulfur centers (using the VWN–Stoll potential). These centers are antiferromagnetically coupled and described using BS DFT. The ligands were simplified for the computational studies. Metal–ligand bond lengths and angles are very well described, with average errors of less than 0.05 Å, ligand bridge angle errors less than 10°, and M–M distance errors from 0.02 to 0.08 Å. Geometry variations with change in oxidation state or protonating oxo bridges are well portrayed. For two complexes, we studied the geometric consequences of spin coupling in greater detail by using both the high-spin and BS states to generate the spin ground-state energy (here total S = 0) as a function of geometry. Equivalently, the Heisenberg J is treated as a function of geometry J(x). The predicted S = 0 M–M distance shortens further, by about 0.1 Å, and the M–M distance error increases. There are probably three contributing factors here. These calculations were conducted in the gas phase, and solvation effects should somewhat expand the Fe–Fe distance in a diferric iron–sulfur dimer complex, and for the Mn–Mn distance in an Mn(IV)2(μ-O)3 2+ complex. The simplification of the terminal bonded ligands should also have a geometric effect. Finally, in many transition metal dimer complexes with LSDA, GGA, or even hybrid HF–GGA potentials, the Heisenberg exchange coupling is predicted to be too strong. Very recently, we examined both the geometry and Heisenberg exchange coupling in a dinuclear Mn(III)Mn(IV) complex with the full macrocyclic ligands in collaboration with Neese’s group using the ORCA and ADF quantum chemistry codes [71]. The predicted geometry by either B88P86 or B3LYP was excellent, with B3LYP a little more expanded. B3LYP gave much better predictions for the Heisenberg J coupling compared with experiment in this case, consistent with overestimation of metal–metal interaction energy with B88P86.
Iron-oxo dimer enzymes: active-site structures, energetics, and properties
In iron-oxo- (hydroxo-) bridged dimer enzymes, MMOH and RNR, we have found that the PW91 potential for both exchange and correlation provides high-quality geometries [72, 73]. In the gas phase, very reasonable Heisenberg coupling parameters are calculated, but geometry optimization in a solvent can give too-strong antiferromagnetic coupling in some cases [74]. Since most properties are strongly improved with solvation calculations, we can conclude that the error in spin coupling is intrinsic to this exchange–correlation potential. With a spin Hamiltonian, H spin = −2J S 1 · S 2 , the calculated J parameters for MMOH(oxidized) and MMOH(reduced) [Fe(III)2 and Fe(II)2] are −35 cm−1 (−39 cm−1 in solvent) and +32 cm−1 (+5 cm−1 in solvent) compared with experiment, −4 to −10 and +0.4 cm−1. For RNR(ox) and RNR(red) [Fe(III)2 and Fe(II)2], J(calc.) = −130 cm−1 (−240 cm−1 in solvent) and +13 cm−1 (−6 cm−1 in solvent) compared with J(exp) = −90 to −108 cm−1 (oxidized), and −0.5 cm−1 (reduced). As we have observed before in other transition metal dimer and tetramer complexes (manganese-oxo and iron–sulfur complexes) [56, 75–78], there is a consistent and correct trend with changes in oxidation state, and with the protonation state of the bridging ligands, but J parameters are not quantitatively reliable. We note that a difference in J of 100 cm−1 for a diferric complex with high-spin sites is chemically equivalent to a difference in E(HS) − E(S = 0) of about 8 kcal mol−1, or 5 kcal mol−1 of “spin-bonding” energy for E(S = 0) with respect to the “spin-non-bonding” spin barycenter.
Evaluating the relative spin state energetics for Fe(III) (S = 5/2, 3/2, or 1/2) and Fe(IV) (S = 2 or 1) sites in these enzymes requires some care, but the connection to experiment can be sorted out by comparing structures, properties, and energetics for various computational models with spectroscopy and experimental structural data [79]. The biological systems have high-spin metal sites throughout the catalytic cycle, and calculated Mössbauer parameters are in much better agreement with experiment when the computational models have high-spin Fe sites. The high oxidation state intermediate of MMOH, intermediate Q [Fe(IV)–L–Fe(IV)] is the active species for the oxidation of methane to methanol [2, 80–82], and intermediate X of RNR [Fe(III)–L–Fe(IV)] oxidizes tyrosine to a stable tyrosine radical [83]. Since there are no X-ray structures for these high-valent intermediates, we tested an extensive set of possible structural models by comparisons with Mössbauer spectroscopy for MMOH(Q) [2, 81], and Mössbauer, electron–nuclear double resonance (ENDOR) [84, 85], and magnetic circular dichroism (MCD) spectroscopy [86, 87] for RNR(X) as well as energetic analysis for valence isomers and protonation states [74, 79, 88]. Our best current structure for MMOH-Q has an Fe(IV)-di-μ-oxo-Fe(IV) structure as anticipated by Shu et al. [80]. Our best current structure is depicted in Fig. 1. It has an Fe(IV)–Fe(IV) center, with each Fe(IV) high spin (S = 2) and antiferromagnetically coupled to yield net spin S = 0. The predicted energy of this state is lower than that for intermediate spins (S = 1) on the two Fe(IV) sites antiferromagnetically coupled, and the predicted Mössbauer quadrupole splittings are better for the high spin–high spin state than for the intermediate spin–intermediate spin state. The high spin–high spin versus intermediate spin–intermediate spin states also have significantly different geometries, with a very short Fe–Fe distance for intermediate spin–intermediate spin, 2.42 Å, while high spin–high spin has a longer Fe–Fe distance, 2.63 Å. Our model is related to a model proposed by Siegbahn [89], but is more open at the iron sites. Another large structure, related to that proposed by Baik et al. [82] is shown in Fig. 2. This model is currently being tested with additional second-shell waters.

Our current best model for the active site of methane monooxygenase intermediate Q (MMOH-Q). (Reproduced with permission from [2]. Copyright 2004 American Chemical Society)

By contrast, for RNR-X, our best model, Fig. 3, which fits most spectroscopic data very well, differs from those proposed by other groups which we have tested. We propose an Fe(III)-di-μ-oxo-Fe(IV) core for this structure. Siegbahn has proposed two different models based on an Fe(IV)-μ-oxo-μ-hydroxo-Fe(III) core; his second model is similar to ours, but it contains the protonated (OH−) bridge, and also corresponds to a different valence isomer for the Fe(III) and Fe(IV) sites with respect to the active tyrosine. In our proposed structure, both μ-oxo ligands are derived from O2, and we propose (Fig. 4) that a proton that enters at the step between the diferric-peroxo and the next state that we call Pre-X(t) acts as a catalytic proton for peroxo reduction and bond breaking, and leaves the center when intermediate X is finally formed. This also rationalizes some observations of Hoffman’s group for 17O and proton ENDOR that are not consistent with other structures and mechanisms. The most effective calculations (we employed PW91 with ADF) use either very large complexes (as in MMOH-Q) with first- plus second- and some third-shell ligands for geometry optimization or smaller models (for RNR-X) geometry-optimized with a continuum solvation model. While the geometries are typically improved after including solvation (with a conductor-like screening model, COSMO), this can be difficult to establish when the comparison is with medium-resolution X-ray protein structures or without direct structural information available except by extended X-ray absorption fine structure (EXAFS). More definitively, calculated Mössbauer parameters (both isomer shifts and quadrupole splittings) are often dramatically improved compared with those from experimental Mössbauer spectra. For fitting Mössbauer isomer shifts, it is very important that the fitting “test set” of synthetic iron complexes also be geometry-optimized in solvent.

Our best model for the active site of ribonucleotide reductase intermediate X (RNR-X) [79]. Labels are for RNR from Escherichia coli

Feasible path showing how RNR-X is formed by reaction of O2 with the reduced RNR-R2 diiron center. (Reprinted with permission from [79]. Copyright 2005 American Chemical Society)
In some cases, the inclusion of observed structural waters in the active site can make a dramatic difference both for calculated energetics and for properties. We have examined the active site of oxidized diferric MMOH, where medium-resolution X-ray structures are available for two different bacteria (Methyosinus tricosporium and Methylococcus capsulatus). A significant question is whether the bridging solvent ligands are [1(OH)−, 1(H2O)] [90] or 2(OH−) [91]. One of the X-ray structures (for Methyosinus tricosporium) contains a cluster of four waters forming a hydrogen-bond network to the bridging solvent in question and including other metal-bound hydroxyls, waters, and glutamates. With DFT methods, with a PW91 potential, the calculated pK a of this bridging water changes from 0.1 to 5.4 when these discrete waters are included in the quantum cluster, and optimization is done including also continuum COSMO solvation; the Mössbauer properties are also greatly improved [74]. In the Methylococcus capsulatus X-ray structure, there is good evidence that the “extra proton” is retained in the bridge, giving [1(OH)−, 1(H2O)], but this state is stabilized strongly by a second-shell exogenous acetate from the crystallization buffer. In Methyosinus tricosporium, the water cluster replaces this acetate, and the 2(OH−) form is more energetically stable.
For the optical transition energies of RNR(X) used for the MCD analysis mentioned before, we employed ΔSCF methods and the related Slater transition state approach [44] to improve the stability of excited hole states. This was used for Fe(IV) site d → d excitation calculations [87, 88]. The environment was modeled as a high dielectric constant aqueous continuous medium with a combination of the COSMO for geometry optimization, and subsequent vertical self-consistent-reaction-field methods for these single-electron excitations. These methods should be comparable (or better) in quality to time-dependent DFT for d → d excitations. In previous work [62, 92], we found that ΔSCF (or its Slater transition state approximation) gave good-quality excitation energies for ligand to metal charge transfer excitations in both cobalt semiquinone valence isomeric complexes and in iron–sulfur dimer complexes. For the iron–sulfur dimer complex (reduced state) Fe(II) d → d excitations were of good accuracy using the Slater transition state. For a Co(II)(semiquinone)2(phenanthroline) complex, one-electron orbital energy differences were sufficient to predict the low-lying metal–ligand charge transfer and ligand field (d → d) bands.
Coupled electron and proton transfer in manganese superoxide dismutase
SODs catalyze the dismutation of superoxide radical ions with proton binding to give hydrogen peroxide and molecular oxygen. The net reaction is
In combination with catalase, which converts product hydrogen peroxide to molecular oxygen and water, SODs protect living cells from toxic oxygen metabolites formed as part of both normal and pathological metabolic pathways, for example, in the electron transport chain in mitochondria. MnSOD is the SOD protein isoform in the mitochondrial matrix, where it evidently functions to dismute O •−2 generated when O2 gains access to reduced factors in the electron transport chain. (There are similar MnSODs in some bacteria, and in others there are FeSODs which operate by a similar catalytic cycle.) O •−2 production occurs in both complex I (NADH-UQ oxidoreductase) and complex III (bc 1 complex). Complex III contains the Rieske FeS protein and other metalloprotein centers, to be discussed later.
We focus on the coupling of electron to proton transfer in oxidized Mn3+SOD. There are important kinetics issues here, and the redox energetics are also very informative. If we let M3+(OH)− represent the oxidized MnSOD active site containing a bound hydroxyl ion and M2+(OH2) represent the same site for the reduced enzyme with a bound water (Figs. 5, 6), the half reactions for the two halves of the catalytic cycle are

Active site of Fe and Mn superoxide dismutases (SODs). Labels are for wild-type human MnSOD. (Reprinted with permission from [2]. Copyright 2004 American Chemical Society)

Catalytic cycle of the dismutation of superoxide ion via alternating reduction of the M3+SOD (M is Mn or Fe) and oxidation of the M2+SOD enzyme. (Reproduced with permission from [5]. Copyright 2002 American Chemical Society)
Notice that each half of the catalytic cycle involves an O •−2 binding step and a subsequent proton binding step to give the products O2 and H2O2. The successive protonation of the M3+-bound OH− and deprotonation of M2+(OH2) with associated electron transfer in successive halves of the catalytic cycle was proposed by Lah et al. [17], and followed the kinetics study of FeSOD by Bull and Fee [18] where they showed that FeSOD takes up one proton upon reduction. Our combined DFT/electrostatics calculations (B88P86 exchange–correlation potential) of the pK a of M3+–H2O and M2+(OH2) enzyme active sites showed these are strongly acidic and strongly basic, respectively, and provided strong support for this model. The more conventional and widely used approach is to ignore the active-site protonation in the first of the two equations, and to put 2H+ into the second equation. This destroys a proper interpretation of the kinetics in the cycle of Fig. 6. Further, the fundamentally important redox potential for this catalytic cycle is
describing the coupled electron and proton transfer to the active metal site. From Fig. 6, it is clear that the rate constant k 2 governing the release of O2 from the active site after O −2 binding is linked with prior or simultaneous binding of a proton to M3+(OH)− as before. The rate constant for k 2 (per second) is fairly low and can be lowered even more by mutation of the native Tyr34 or Gln143 residues in the wild-type human enzyme to Phe34 or Asn143, respectively. These two residues form a hydrogen-bonded network to the active-site Mn-bound OH− (Fig. 5). Our analysis indicates that upon O •−2 binding to the active site, there is partial negative charge transfer to the Mn3+ ion, but full one-electron transfer is prevented until a proton is transferred to generate H2O from OH−. The partial negative charge transfer facilitates the proton transfer through the hydrogen-bonded network from Tyr34 to Gln143 to the Mn3+-bound OH−, but this requires inner-sphere binding of O •−2 . It is difficult to see how longer-range electron transfer via tunneling could promote the proton transfer; probably these two steps would become energetically decoupled in this case.
In MnSOD and FeSOD as well as CuZnSOD [6], the consensus mechanism currently implicates inner-sphere binding of O •−2 in both halves of the catalytic cycle. (The situation for NiSOD is not so clear [93–96].) However, there are many metalloprotein complexes where electron transfer occurs by tunneling, for example, in electron transport chains where electrons are passed between iron–sulfur proteins and cytochromes (typical distances are 11 Å), in the multielectron–multiproton reduction of the nitrogenase system (involving binding and electron transfer from the Fe protein into the FeMo protein and eventual electron/proton transfer to the FeMo cofactor center), or the electron transport pathway from the RNR subunit 2 tyrosine radical into the subunit 1 reaction site (34 Å away). Some of these redox events are clearly linked to proton transfer. Can proton coupling be linked in a single energetic step to electron transfer if the electron transfer occurs by tunneling? Alternatively, if the electron is transferred to a delocalized orbital, or if the electron transfer has strongly associated electron relaxation so that the net charge change is spread over a large volume, then decoupling the proton transfer may not be so energetically costly.
Returning to the MnSOD and FeSOD redox potentials, it has proven extremely difficult to measure the redox potential for M3+(OH)− + H+ + e − → M2+(OH2) directly via electrochemistry. There may be many contributing reasons, but our favorite is that outer-sphere electron transfer in MnSOD and FeSOD is very difficult for the reason given before; inner-sphere electron transfer couples much more efficiently to proton transfer. The best way then to measure the redox potential is from the kinetics of the small inner-sphere electron donor superoxide anion itself. From experimental kinetics rate constants, we obtain redox potentials for MnSOD for coupled electron transfer/proton transfer from bacterial Thermus thermophilus, human wild type, and human Q143N of 0.40, 0.32, and 0.59 V. This is the same ordering as obtained from DFT/electrostatics methods: −0.25, −0.29, and −0.11 V. The absolute error from the DFT redox calculations is about 0.6 V (or 14 kcal mol−1). For FeSOD, our error is smaller: 0.25 V, from experiment at pH 7 versus 0.16 V from DFT. As calibration, earlier we computed the redox potential for Fe3+,2+ and Mn3+,2+ redox couples in aqueous solution at pH 7, where there is a coupled one-electron, one-proton transfer. For Fe3+,2+, the results are 0.41 V (DFT) versus 0.48 V (experiment), and for Mn3+,2+, 0.79 V (DFT) versus 1.15 V (experiment). The errors for the M ions in aqueous solution account for almost all the error in Fe3+,2+SOD and about half the error in Mn3+,2+SOD. Some of the redox error in our DFT/electrostatics calculations for MnSOD probably arises from our neglect of the π-cation interaction between the tryptophan ring and the Mn coordination shell, since either Mn3+–OH−–Asp− or Mn2+–OH2–Asp− has a net +1 charge to interact with the tryptophan π system. Taking the π-cation interaction into account would require enlarging the quantum model of Fig. 5 to include the indole ring.
Iron–sulfur metalloenzymes
Rieske center redox and pK a
Complex III contains the Rieske FeS protein and a number of cytochromes (b L, b H, and c 1). The interaction of the mobile electron and proton carrier molecule, reduced ubiquinone (UQH2), with the Rieske center generates a ubisemiquinone radical (UQ•−), with one electron being passed from the Rieske center to cytochrome c 1 and then on to the mobile protein cytochrome c, and two protons. The two protons eventually pass into solution on the P-side (cytoplasmic side of the inner mitochondrial membrane). While the detailed proton path is not known, there is now good evidence that two protons are bound [97] to the Rieske center after one-electron reduction at physiological pH, but fewer protons are bound in the oxidized state. Meanwhile the UQ•− to UQ (oxidized quinone) couple is near to the O •−2 to O2 couple in redox potential, which allows production of superoxide in some cases.
The Rieske iron–sulfur complex center is shown in Fig. 7. The X-ray structure of the Rieske fragment proteolytically cleaved from its membrane anchor from the bovine cytochrome bc 1 complex was determined to 1.5-Å resolution [98]. This was used as a starting structure for geometry optimization of the large quantum model complex in Fig. 7. As mentioned previously, the Rieske center is involved in the oxidation of reduced ubiquinone (UQH2) [22]. The Rieske iron–sulfur protein becomes reduced and protonated by one or two protons. Usually, the one-electron-reduced Rieske center has two protons bound, one on each Fe(II)-bound histidine, while the oxidized Rieske center has zero, one, or two protons bound (or a statistical mixture of these) depending on pH. (Two protons are bound at very acid pH and zero at very basic pH in the oxidized Rieske center, while in the reduced Rieske center only very high pH allows for some deprotonation.) The electron is then passed to cytochrome c 1 and the protons are presumably transferred out to the cytoplasmic P-side of the inner mitochondrial membrane, although many aspects of this remain unclear. Our DFT calculations using a PW91 potential and a large cluster model plus protein/solvent provided strong evidence in favor of coupled one-electron transfer with histidine protonation at both His141 and His161 [97, 99]. The apparent pK as calculated for the oxidized state (6.9, 8.8) are in good agreement with those measured (7.5, 9.2) for the bovine Rieske fragment [100] and for Rieske from T. Thermophilus (7.85, 9.65) [97]. On one-electron reduction, the pK as shift to (11.3, 12.8), in good agreement with the experiments of Link et al. [100] for bovine Rieske center (more than 10) and for T. Thermophilus (about 12.5) [97]. Further the calculated redox potential of −12 mV at the acid limit is reasonable compared with the experimental values of +311 mV (bovine) and +161 mV (T. Thermophilus) For the alkaline limit, E 0 = −500 mV (calculated) versus −275 mV (experimental, T. Thermophilus). Spectroscopic analysis of the reduced electronic state is considered next.

Structure of the Rieske iron–sulfur complex center. (Reprinted from [99] with kind permission of Springer Science and Business Media)
Rieske electron paramagnetic resonance spectra, spin coupling, and delocalization
In very recent work from the Munck and Fee groups [101], they found that the electron paramagnetic resonance of the Rieske 2Fe2S protein in the reduced [2Fe2S]+ state (spin doublet S = 1/2) is extremely anisotropic at pH 14, with g values of 1.81, 1.94, and 2.14 compared with typical g values of 1.80, 1.90, and 2.02 for pH 7. Such highly anisotropic g values occur in some other 2Fe2S ferredoxins as well, in particular in some systems and states (called signal II) of the xanthine oxidase class [102]. The analysis briefly presented here opens a whole new window into the physical implications of these highly anisotropic g tensors.
In the spin Hamiltonian, only an antisymmetric exchange term has been shown to be able to produce this large anisotropy. We will not deal with the full physics of antisymmetric exchange, except to say that this involves both exchange coupling and spin–orbit coupling operating together. For an antisymmetric spin Hamiltonian of the form d · S 1 × S 2 , the g tensor depends on the ratio of the antisymmetric vector parameter d (x component) |d x | to the effective Heisenberg J parameter, |d x |/J. A proper fit to the experimental g tensor requires an anomalously low Heisenberg exchange coupling parameter J (in the spin Hamiltonian term H = J S 1 · S 2 ) at pH 14, J = 43 ± 10 cm−1 (positive, antiferromagnetic) while typically J > 150 cm−1 for reduced ferredoxin states. Equivalently, the spin quartet state (S = 3/2) is very low lying, but why? What does this tell us about the energetics at the reduced site? We now think we have a clear understanding of this issue with important implications. The effective Heisenberg J parameter can be diminished by a ferromagnetic term J F due to incipient (partial) electron delocalization (Robin–Day class II) as described in Blondin and Girerd [64] and Noodleman et al. [69] (see also Bominaar et al. [65]). This term takes the form J F = −2B 2/ΔE, where B is the double exchange or resonance delocalization parameter, and ΔE = E optical = ΔE(vibronic) + ΔE AB, where ΔE is the site trapping energy, which is the sum of a vibronic energy plus the site inequivalence energy ΔE AB [this is the energy difference between reduction at the Fe(A) adjacent to two histidines, and Fe(B) adjacent to two cysteines; reduction at Fe(B) is higher in energy than at Fe(A), and ΔE AB = E B − E A]. From our previous calculations (Ullmann et al. [99], Table 4), changing the pH from 7 to 14 strongly reduces ΔE AB from about 9 to nearly 0 kcal mol−1 (from 3,100 cm−1 to about 100 cm−1) by deprotonating both histidine ligands to Fe(A). [Typical values for B = 600–900 cm−1 and ΔE(vibronic) = 4,000 cm−1 are found from calculations and spectroscopy.] Taken together, these will strongly increase the magnitude of J F and lower the total J by decreasing ΔE. The overall effect has the correct magnitude as well. In this and other systems, both DFT/electrostatics calculations and modeling can be used to explore the effects of pH and active-site geometry or protein environmental effects on ΔE. ΔE AB has thermodynamic and structural implications, while total ΔE is a reorganization energy, which can affect electron transfer kinetics. Further, ΔE = E optical is the energy of the optical intervalence charge transfer band, the intensity of which is weakened by spin coupling, and is therefore often difficult to measure directly. Reductions in J due to a low value of ΔE AB may occur in some ferredoxins, but this effect depends on the Fe site asymmetry induced by the protein–solvent environment as well as on the intrinsic asymmetry within the active-site complex.
To cant or cant not: electronic structure of oxidized high-potential 4Fe4S proteins
4Fe4S proteins operate between two different redox couples, Fe4S4(SCys)4 −,2−, called the “high-potential” couple, and Fe4S4(SCys)4 2−,3−, called the “ferredoxin couple”. The proteins operating with the “high-potential” couple in their normal electron transfer operation are distinct from ferredoxin proteins. Further, it is known that 4Fe4S ferredoxins are often unstable to loss of one or more Fe atoms or to cluster disintegration in their superoxidized 1− state, while “high-potential” 4Fe4S proteins (HIPIP) readily redox cycle between 1− and 2− states and are stable in the 1− form. Very recently, J. Fee’s group at Scripps (including L. Hunsicker-Wang and D. Stout) obtained new high-resolution X-ray structures for oxidized and reduced (1.35 and 0.92 Å, respectively) HIPIP from T. tepidum. Further, they analyzed several different HIPIP X-ray structures from the literature comparing oxidized and reduced HIPIPs with the analogous (2−) oxidized form of ferredoxin proteins. The HIPIPs are structurally different from the ferredoxin oxidized in both the HIPIP 1− and the HIPIP 2− states. The HIPIPs (both oxidized and reduced) consistently contain one short nonbonded Sγ–Sγ distance at about 5.95 Å (for the coordinated SCys groups), while the other five Sγ–Sγ distances are longer, in the range 6.2–6.5 Å. The latter distances resemble those in ferredoxins (2−).
This structural analysis was facilitated by earlier collaborative work between our groups [103], where the interlaced irregular tetrahedra of the irons, bridging sulfurs, and terminal sulfurs are each inscribed in a different sphere, with a different center, allowing analysis of angular distortions from regular tetrahedra. This “circumsphere” analysis is based on an old, but highly nontrivial theorem from solid geometry. This approach allows both geometric and energetic analysis of radial and angular distortions, combining DFT calculations with X-ray crystallography for structures.
Working with the Fee group, we have uncovered a consistent electronic explanation for this structural difference between HIPIPs and ferredoxins. There are three electronic states possible for the oxidized (1−) HIPIP, called OS1, OS2, and OS3 [77, 104]. OS1 and OS2 mix in low protein type symmetries, but OS3 is distinctive. Electronically, they differ as follows: OS3 contains a delocalized mixed-valence Fe2+–Fe3+ pair coupled to pair spin S 34 = 9/2, while the other pair, Fe3+–Fe3+, contains high-spin ferric ions, which are spin-canted to give pair spin S 12 = 4, and total spin S = 1/2. The electronic states OS1 and OS2 yield the same total spin S = 1/2, and have the same spin for the mixed-valence pair S 34 = 9/2, but S 12 = 4 is achieved in a different way. Here spin crossover combined with spin-forbidden charge transfer character yields a site spin of S 1 = 3/2 which combines with high-spin S 2 = 5/2 to give S 12 = 4. More precisely, the high-spin and intermediate-spin sites S 2 and S 1 can interchange and form a RSCP where S 12 = 4. The HIPIP oxidized geometry with one short Sγ–Sγ distance (at about 5.95 Å) strongly favors OS1 and OS2 over OS3, and this is evidently favored by the HIPIP protein environment since both oxidized and reduced HIPI have similar distortions. There is more SCys to Fe3+ charge transfer character in OS1 and OS2 than in OS3, and the Fe3+–SCys bond polarity is less in the (SCys)2(Fe3+)2(Sbridge)2 layer [8]. We are examining the further implications of these unusual electronic and geometric structures for HIPIP proteins. This probably accounts for the lower reactivity of oxidized (1−) HIPIP than for “superoxidized” (1−) ferredoxin toward atom loss or cluster decomposition and likely modulates the HIPIP redox potential as well.
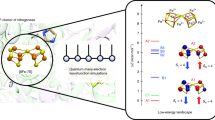
Cluster assembly and central-atom insertion in the FeMo cofactor of nitrogenase
The FeMo cofactor in the nitrogenase MoFe protein is certainly one of the most exotic metal cofactors in biology both structurally and functionally (Fig. 8) [105]. The determination of the X-ray structure of the MoFe protein required many years of work from several laboratories before the first structures became available in 1992– 1993. The initial structures were at 2.2-Å resolution (Rees’s group) [106] and 3.0-Å resolution (Bolin’s group) [107]; the 2.2-Å structure from Rees’s group was later refined to 2.0 Å, and a later structure from Smith’s group was refined to 1.6 Å [108]. All of these structures showed unusual threefold coordination for the six central prismatic Fe atoms, and a hole in the center of the cluster. In 2002, a very high resolution structure from Rees’s group [109] showed that a previously unrecognized ligand resides in the center of the catalytically essential FeMo cofactor. The resulting electron density map shows that the central density is consistent with three possible heavy atoms, carbon, nitrogen, or oxygen, and that sulfur is too large with too-high an electron density to fit the map. The vacancy site in the prior X-ray structure electron density maps is an artifact due to limited resolution. The finite resolution produces an artificial negative electron density which masks the actual positive electron density at the central site; this problem is a consequence also of the presence of six rather symmetrically positioned Fe atoms.

FeMo cofactor of nitrogenase with an unknown ligand X sitting in the center. (Reprinted with permission from [105]. Copyright 2003 American Chemical Society)
It is not at all surprising that the missing heavy atom also diverted the theoreticians, who are known to be very trusting people. We were among a number of groups who explored the structures and properties of FeMo cofactor before Rees’s revised structure was published in September 2002 [110–113]. In these earlier papers, sensible geometric structures for the active-site FeMo cluster could be obtained, but our calculated redox potential (Mox + e − → MN) was far too positive at +820 mV compared with experiment, −42 mV for Azotobacter vinelandii (vs. the standard hydrogen electrode) [111] (Fig. 9). By contrast, the 4Fe4S proteins give very reasonable redox potentials by combined DFT (B88P86) and electrostatics calculations [8]. The average error is about 200 mV too negative compared with experiment, and the slope of the calculated versus experimental redox potentials is nearly ideal (slope of 1) over three different redox couples (1−,2−), (2−,3−), and (3−,4−) and a wide redox range (over 1 V). As seen in [111], we knew that something was wrong for the FeMo cofactor, but what was it? When the existence of the central atom was discovered, we and others rebuilt our models (Fig. 8). The central atom is a central atomic anion, and C4−, N3−, and O2− are the possibilities. For the system with the central hole, our best model for the resting state (called MN, with total spin S = 3/2) contained 6Fe2+1Fe3+ as the Fe oxidation state with E 0 = +820 mV. From the known half-integer spin state of MN, we know that the number of electrons in the cofactor center is odd, so we can focus on two possible oxidation states for MN. Now with a central anion, our best redox potential was obtained for a central N3− and with altered Fe oxidation states 4Fe2+3Fe3+ (Fig. 9), +190 mV [2, 105]. As shown in Fig. 10, neither C4− nor O2− gives decent redox potentials (much too negative and too positive, respectively) for the 4Fe2+3Fe3+ proposed form of MN. As we argued previously, for O2−, the resting state could possibly have Fe oxidation states 6Fe2+1Fe3+ if one of the central μ2S sulfides is protonated. This would give a cluster charge which should lead to a reasonable redox potential, but this leads to a structure on the protonated μ2S sulfide with too-long Fe–S bonds (2.35, 2.38 Å), much longer than in the high-resolution structure (2.21–2.25 Å). Correspondingly, with C4− the expected Fe oxidation states for MN are 4Fe2+3Fe3+, but again one μ2S needs to be protonated to obtain a reasonable redox potential, and there again should be two long Fe–S bonds to the μ2SH. Overall, we judge that our argument is more strongly against O2− than C4− since we have done the structural test. In general, we still favor a central N3− (nitride) as the ligand with oxidation state 4Fe2+3Fe3+ and Mo4+ for MN. Hoffman’s group has developed ENDOR and electron spin echo envelope modulation (ESEEM) evidence against the presence of a central N3− since they see no identifiable central N by 14N, 15N ENDOR, and ESEEM spectroscopy when the FeMo cofactor is extracted from the resting state holo-MoFe protein in N-methylformamide [114]. In this way, all protein bound N signals are lost on cluster extraction. They believe that their experimental setup would allow them to see 14N signals of about 0.1 MHz or less. Instead, they favor a central C4− (carbide). However, our own view is that the opposite spin alignment among the six prismatic Fe atoms may lead to a very low effective spin density on the central N after accounting for spin coupling. A definitive resolution of the central atom still awaits further quantum chemical modeling and further experimental examination.

Calculated FeMo cofactor redox potential versus experiment. For comparison, redox potentials calculated for a range of iron–sulfur proteins are also shown. (Reprinted with permission from [105]. Copyright 2003 American Chemical Society)

Correlation of calculated versus experimental redox potential for the FeMo cofactor. (Reprinted with permission from [105]. Copyright 2003 American Chemical Society)
In any event, either nitride or carbide is unprecedented in biological systems, which leads us to consider how such a cluster with a central multiply charged anion can be formed. We start from what is known from genetic and biochemical analysis, and what has recently been discovered by EXAFS in the work of Corbett et al. [115]. Then we can make a reasonable proposal for a sequence of events based on comparing our calculated redox potentials (Mox + e − → MN) for a central vacancy (hole) using either 6Fe2+1Fe3+ or 4Fe2+3Fe3+ as our model for MN with the redox potential for MN where the central N3− is present and the oxidation state is 4Fe2+3Fe3+ for the cluster. All these models contain Mo4+ and the chelating homocitrate. We consider also the one-electron reduction (MN + e − → MR) in the presence of the central vacancy.
Corbett et al. [115] were able to isolate by genetic manipulation an iron–sulfur cluster containing FeMo cofactor precursor bound to NifEN; the latter is an intermediary assembly protein. (We will call this the “NifEN-bound-precursor” or “NifEN-bp.”) It is important to recognize that the FeMo cofactor is assembled outside the MoFe protein in a stepwise process, and then it is transported and inserted into the apo-MoFe protein. These steps involve the precursor iron–sulfur cluster bound to NifB protein, called NifB-co, which is then transferred to the NifEN protein. Earlier chemical and genetic studies indicated that the Mo and homocitrate are incorporated into the FeMo cofactor later; the EXAFS studies of Corbett et al. provide further support for this. Their best EXAFS fits for Fe K-edge spectra indicate that Mo is absent and that the relevant cluster contains seven or eight Fe atoms (more likely eight). The EXAFS is not sensitive enough to detect the scattering of a small central atom against the background of heavy atom scatterers (Fe and S), so it is unknown whether the central N (or C) is present at this stage.
Our goal is to understand some of the basic steps needed to insert the central ligand into the NifEN-bound precursor, and their consequences. We will assume that the central ligand inserted is nitride (N3−), but the argument will be broadly similar, if carbide (C4−) is the central ligand. (We consider carbide as the more anomalous case on the basis of biochemistry.) At an early stage, the N3− is absent from NifEN-bp. From our previous calculations (Figs. 9, 10), it is clear that the redox potential involving Mox + e − → MN is very high if there is a central vacancy and the MN oxidation state is 4Fe2+3Fe3+. For the intact FeMo cofactor cluster in the protein (including Mo4+ bonded to homocitrate) (Figs. 8, 10, 11), this redox potential is +1,300 mV, and even for the 6Fe2+1Fe3+ state for MN, the corresponding redox potential is +820 mV [111]. Similar redox potentials are calculated in an aqueous solvent environment rather than in the protein (+550–630 mV). For the redox couple MN + e − → MR (or MI) corresponding to 6Fe2+1Fe3+ + e − → 7Fe2+, the relevant redox potentials with the central vacancy are +160 and +150 mV in the MoFe protein and +430 and +370 mV in aqueous solvent. It is therefore very probable that the early NifEN-bp cluster before N3− insertion is in the all-ferrous 7Fe2+ or 8Fe2+ state. [Formally, Mo4+ homocitrate (4−) is neutral, but on coordination to the cluster, two carboxylates are well away from the Mo and screened by a water cluster and by a glutamine in the holoprotein structure; the Fe2+ acting as the substitute for Mo4+ homocitrate likely has a similar charge near the coordination site, as Fe2+(H2O)3, for example.] We propose that a noncatalytic deamination of glutamine or asparagine leads to a successive stripping of protons and carbon from the terminal N, yielding N3−, but this needs to be coupled to successive oxidation of some of the coordinating Fe sites (and perhaps of other Fe sites as well). Overall a conversion of 8Fe2+ → 5Fe2+ + 3Fe3+ + 3e − is likely, so the incorporation of nitride requires an electron sink for oxidation of the forming complex with the central ion. If the μ2S atoms are still in the form of persulfides, then six electrons are needed to yield 3μ2S2− plus sulfides still bound to the NifEN assembly complex. Three electrons could come from the 8Fe cluster and three more from NifEN, but fewer are needed if some μ2S remain as sulfides throughout. Fe2+ persulfides will have weaker Fe–S bonding than Fe2+,3+ sulfide, and will facilitate access to the central vacancy site by nitride. This mechanism will also prevent the danger of further oxidation of Mo4+ homocitrate (4−) to a homocitrate radical (as we observed in our early calculations [110, 111]), so there is good chemical logic for Mo4+ homocitrate (4−) being incorporated late in FeMo cofactor cluster formation (Fig. 11).

Important hydrogen-bonding interaction from the local protein environment with FeMo cofactor. s side-chain-based interaction with FeMo cofactor; b backbone-based interaction. (Reprinted from [111] with kind permission of Springer Science and Business Media)
Concluding remarks
In this review, we have ranged widely, covering some of the fundamentals of DFT, and selected applications. By combining structural, energetic, and spectroscopic calculations with experimental data, considerable insights can be derived for many electronic states and redox-dependent catalytic and activation pathways. We have also seen that properties evaluation strengthens mechanistic and electronic assignments that would be difficult considering energies alone.
Notes
We warn the reader at this point that the terms “electron exchange”, or “electron exchange potential,” or “exchange hole” have different meanings in HF and DFT methods because these methods are constructed from different starting points. There is a fairly close correspondence for atoms, but not for molecules, and these differences are important.
Abbreviations
- ADF:
-
Amsterdam Density Functional
- BP:
-
Becke 1988–Perdew 1986
- BS:
-
Broken symmetry
- B86:
-
Becke 1986
- B88:
-
Becke 1988
- COSMO:
-
Conductor-like screening model
- DFT:
-
Density functional theory
- ENDOR:
-
Electron–nuclear double resonance
- ESEEM:
-
Electron spin echo envelope modulation
- EXAFS:
-
Extended X-ray absorption fine structure
- GGA:
-
Generalized gradient approximation
- G96:
-
Gill 1996
- HF:
-
Hartree–Fock
- HIPIP:
-
High-potential 4Fe4S protein
- LSDA:
-
Local spin density approximation
- MCD:
-
Magnetic circular dichroism
- MM:
-
Molecular mechanics
- MMOH:
-
Methane monooxygenase
- PW91:
-
Perdew–Wang 1991
- QM:
-
Quantum mechanics
- RNR:
-
Ribonucleotide reductase
- RSCP:
-
Resonance spin crossover pair
- SCF:
-
Self-consistent field
- SOD:
-
Superoxide dismutase
- VWN:
-
Vosko–Wilk–Nusair
References
Frausto da Silva JJR, Williams RJP (1991) The biological chemistry of the elements. Clarendon, Oxford
Noodleman L, Lovell T, Han W-G, Li J, Himo F (2004) Chem Rev 104:459–508
Miller AF, Sorkin DL (1997) Comments Mol Cell Biophys 9:1–48
Yiklimaz E, Xie J, Brunold TC, Miller A-F (2002) J Am Chem Soc 124:3482–3483
Han W-G, Lovell T, Noodleman L (2002) Inorg Chem 41:205–218
Konecny R, Li J, Fisher CL, Dillet V, Bashford D, Noodleman L (1999) Inorg Chem 38:940–950
Beinert H, Holm RH, Munck E (1997) Science 277:653–659
Torres RA, Lovell T, Noodleman L, Case DA (2003) J Am Chem Soc 125:1923–1936
Asthagiri D, Dillet V, Liu T, Noodleman L, Van Etten RL, Bashford D (2002) J Am Chem Soc 124:10225–10235
Asthagiri D, Liu T, Van Etten RL, Noodleman L, Bashford D (2004) J Am Chem Soc 126:12677–12684
Schueler-Furman O, Wang C, Bradley P, Misura K, Baker D (2005) Science 310:638–642
Hellinga H (1996) Curr Opin Biotechnol 7:437–441
Brazeau BJ, Lipscomb JD (2000) Biochemistry 39:13503–13515
Li J, Nelson MR, Peng CY, Bashford D, Noodleman L (1998) J Phys Chem A 102:6311–6324
Ludwig ML, Ballou DP, Noodleman L (2001) In: Wieghardt K, Huber R, Poulos T, Messerschmidt A (eds) Handbook of metalloproteins. Wiley, Chichester, pp 652–667
Miller A-F, Padmakumar K, Sorkin DL, Karapetian A, Vance CK (2003) J Inorg Biochem 93:71–83
Lah MS, Dixon MM, Pattridge KA, Stallings WC, Fee JA, Ludwig ML (1995) Biochemistry 34:1646–1660
Bull C, Fee JA (1985) J Am Chem Soc 107:3295–3304
Bull C, Niederhoffer EC, Yoshida T, Fee JA (1991) J Am Chem Soc 113:4069–4076
Hsieh Y, Guan Y, Tu C, Bratt PJ, Angerhofer A, Lepock JR, Hickey MJ, Tainer JA, Nick HS, Silverman DN (1998) Biochemistry 37:4731–4739
Chen K, Hirst J, Camba R, Bonagura CA, Stout CD, Burgess BK, Armstrong FA (2000) Nature 405:814
Nicholls DG, Ferguson SJ (2002) Bioenergetics 3. Academic, San Diego
Morales R, Chron M-H, Hudry-Clergeon G, Petillot Y, Norager S, Medina M, Frey M (1999) Biochemistry 38:15764–15773
Fontecilla-Camps JC, Ragsdale SW (1999) Adv Inorg Chem 47:283–333
Greene SH, Richards NGJ (2004) Inorg Chem 43:7030–7041
Kozlowski PM (2001) Curr Opin Chem Biol 5:736–743
Ogliaro F, Cohen S, Filatov M, Harris N, Shaik S (2000) Angew Chem Int Ed Engl 39:3851–3855
Dickerson LD, Sauer-Masarwa A, Herron N, Fendrick CM, Busch DH (1993) J Am Chem Soc 115:3623–3626
Dickerson RE, Geis I (1983) Hemoglobin: structure, function, evolution and pathology. Cummings, Menlo Park
Gilchrist ML Jr, Ball JA, Randall DW, Britt RD (1995) Proc Natl Acad Sci 92:9545–9549
Liu KE, Lippard SJ (1995) Adv Inorg Chem 42:263–289
Stubbe J, van der Donk WA (1995) Chem Biol 2:793–801
Brunold TC, Solomon EI (1999) J Am Chem Soc 121:8277–8287
Hwang J, Krebs C, Huynh BH, Edmondson DE, Theil EC, Penner-Hahn JE (2000) Science 287:122–125
Schultz NE, Zhao Y, Truhlar DG (2005) J Phys Chem A 109:4388–4403
McWeeny R, Sutcliffe BT (1976) Methods of molecular quantum mechanics. Academic, London
Koch W, Holthausen MC (2001) A chemist’s guide to density functional theory. Wiley-VCH, Weinheim
Noodleman L, Lovell T, Han W-G, Liu T, Torres RA, Himo F (2003) In: Lever AB (ed) Comprehensive coordination chemistry II, from biology to nanotechnology. Fundamentals, vol 1. Elsevier, Amsterdam, pp 491–510
Li J, Noodleman L, Case DA (1999) In: Solomon EI, Lever ABP (ed) Inorganic electronic structure and spectroscopy. Methods, vol 1. Wiley, New York, pp 661–724
Handy NC, Cohen AJ (2001) Mol Phys 99:403–412
Schultz NE, Zhao Y, Truhlar DG (2005) J Phys Chem A 109:11127–11143
Xu X, Goddard WA III (2004) J Chem Phys 121:4068–4082
Gill PMW (1996) Mol Phys 89:433–445
Slater JC (1972) Adv Quantum Chem 6:1
te Velde G, Bickelhaupt FM, Baerends EJ, Fonseca Guerra C, van Gisbergen SJA, Snijders JG, Ziegler T (2001) J Comput Chem 22:931–967
Xu X, Goddard WA III (2004) Proc Natl Acad Sci USA 101:2673–2677
Edgecombe KE, Becke AD (1995) Chem Phys Lett 244:427–432
Gritsenko OV, Ensing B, Schipper PRT, Baerends EJ (2000) J Phys Chem A 104:8558–8565
Gritsenko OV, Schipper PRT, Baerends EJ (1997) J Chem Phys 107:5007–5015
Tschinke V, Ziegler T (1990) J Phys Chem 93:8051
Slater JC (1954) Phys Rev 91:528
Noodleman L, Norman JG Jr (1979) J Chem Phys 70:4903–4906
Noodleman L, Post D, Baerends EJ (1982) Chem Phys 64:159–166
Jonkers G, de Lange CA, Noodleman L, Baerends EJ (1982) Mol Phys 46:609–620
Noodleman L, Case DA (1992) Adv Inorg Chem 38:423–470
Zhao XG, Richardson WH, Chen J-L, Li J, Noodleman L, Tsai H-L, Hendrickson DN (1997) Inorg Chem 36:1198–1217
Swart M, Groenhof AR, Ehlers AW, Lammertsma K (2004) J Phys Chem A 108:5479–5483
Deeth RJ, Fey N (2004) J Comput Chem 25:1840–1848
Groenhof AR, Swart M, Ehlers AW, Lammertsma K (2005) J Phys Chem A 109:3411–3417
Anderson PW, Hasegawa H (1955) Phys Rev 100:675–681
Zener C (1951) Phys Rev 82:403
Noodleman L, Baerends EJ (1984) J Am Chem Soc 106:2316–2327
Girerd JJ (1983) J Chem Phys 79:1766–1775
Blondin G, Girerd JJ (1990) Chem Rev 90:1359–1376
Bominaar EL, Borshch SA, Girerd JJ (1994) J Am Chem Soc 116:5362–5372
Middleton P, Dickson DPE, Johnson CE, Rush JD (1978) Eur J Biochem 88:135–141
Bobrik MA, Hodgson KO, Holm RH (1977) Inorg Chem 16:1851–1858
Aizman A, Case DA (1982) J Am Chem Soc 104:3269–3279
Noodleman L, Case DA, Mouesca J-M, Lamotte B (1996) J Biol Inorg Chem 1:177–182
Li J, Noodleman L (1998) In: Solomon EI, Hodgson KO (eds) Spectroscopic methods in bioinorganic chemistry. ACS symposium series 692. American Chemical Society, Washington, pp 179–197
Sinnecker S, Neese F, Noodleman L, Lubitz W (2004) J Am Chem Soc 126:2613–2622
Lovell T, Li J, Noodleman L (2001) Inorg Chem 40:5251–5266
Lovell T, Li J, Noodleman L (2002) J Biol Inorg Chem 7:799–809
Han W-G, Liu T, Lovell T, Noodleman L (2006) J Comput Chem (in press)
Noodleman L, Li J, Zhao XG, Richardson WH (1997) In: Springborg M (ed) Density functional methods in chemistry and materials science. Wiley, New York, pp 149–188
Jordanov J, Roth EKH, Fries PH, Noodleman L (1990) Inorg Chem 29:4288–4292
Mouesca J-M, Chen JL, Noodleman L, Bashford D, Case DA (1994) J Am Chem Soc 116:11898–11914
Mouesca J-M, Noodleman L, Case DA (1995) Int J Quantum Chem Quantum Biol Symp 22:95–102
Han W-G, Liu T, Lovell T, Noodleman L (2005) J Am Chem Soc 127:15778–15790
Shu L, Nesheim JC, Kauffmann K, Munck E, Lipscomb JD, Que L Jr (1997) Science 275:515–518
Lovell T, Han W-G, Liu T, Noodleman L (2002) J Am Chem Soc 124:5890–5894
Baik M-H, Newcomb M, Friesner RA, Lippard SJ (2003) Chem Rev 103:2385–2419
Richard P, Ehrenberg A (1983) Science 221:514–519
Willems JP, Lee HI, Burdi D, Doan PE, Stubbe J, Hoffman BM (1997) J Am Chem Soc 119:9816–9824
Burdi D, Willems JP, Riggs-Gelasco PJ, Antholine WE, Stubbe J, Hoffman BM (1998) J Am Chem Soc 120:12910–12919
Mitic N, Saleh L, Schenk G, Bollinger JM Jr, Solomon EI (2003) J Am Chem Soc 125:11200–11201
Han W-G, Liu T, Lovell T, Noodleman L (2006) (submitted)
Han W-G, Liu T, Lovell T, Noodleman L (2006) J Inorg Biochem 100:771–779
Siegbahn PEM (2001) J Biol Inorg Chem 6:27
Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ (1995) Chem Biol 2:409–418
Elango N, Radhakrishnan R, Froland WA, Wallar BJ, Earhart CA, Lipscomb JD, Ohlendorf D (1997) Protein Sci 6:556–568
Adams DM, Noodleman L, Hendrickson DN (1997) Inorg Chem 36:3966–3984
Szilagyi RK, Bryngelson PA, Maroney MJ, Hedman B, Hodgson KO, Solomon EI (2004) J Am Chem Soc 126:3018–3019
Fiedler AT, Bryngelson PA, Maroney MJ, Brunold TC (2005) J Am Chem Soc 127:5449–5462
Pelmenschikov V, Siegbahn PEM (2006) J Am Chem Soc (in press)
Barondeau DP, Kassmann CJ, Bruns CK, Tainer JA, Getzoff ED (2004) Biochemistry 43:8038–8047
Zu Y, Fee JA, Hirst J (2001) J Am Chem Soc 123:9906–9907
Iwata S, Saynovits M, Link TA, Michel H (1996) Structure 4:567–579
Ullmann GM, Noodleman L, Case DA (2002) J Biol Inorg Chem 7:632–639
Link TA, Hagen WR, Pierik AJ, Assmann C, von Jagow G (1992) Eur J Biochem 208:685–691
de Oliviera FT, Bominaar EL, Hirst J, Fee JA, Munck E (2004) J Am Chem Soc 126:5338–5339
Caldeira J, Belle V, Asso M, Guigliarelli B, Moura I, Moura JJG, Bertrand P (2000) Biochemistry 39:2700–2707
Fee JA, Castagnetto JC, Case DA, Noodleman L, Stout CD, Torres RA (2003) J Biol Inorg Chem 8:519–526
Noodleman L, Peng CY, Case DA, Mouesca J-M (1995) Coord Chem Rev 144:199–244
Lovell T, Liu T, Case DA, Noodleman L (2003) J Am Chem Soc 125:8377–8383
Howard JB, Rees DC (1996) Chem Rev 96:2965–2982
Bolin JT, Campobasso N, Muchmore SW, Morgan TV, Mortenson LE (1993) In: Steifel EI, Coucoucanis D, Newton WE (eds) Molybdenum enzymes, cofactors, and model systems. American Chemical Society, Washington, pp 186–195
Mayer SM, Lawson DM, Gormal CA, Roe SM, Smith BE (1999) J Mol Biol 292:871–891
Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC (2002) Science 297:1696
Lovell T, Li J, Liu T, Case DA, Noodleman L (2001) J Am Chem Soc 123:12392–12410
Lovell T, Li J, Case DA, Noodleman L (2002) J Biol Inorg Chem 7:735–749
Rod TH, Norskov JK (2000) J Am Chem Soc 122:12751–12763
Stavrev KK, Zerner MC (1998) Int J Quantum Chem 70:1159–1168
Yang T-C, Maeser NK, Laryukhin M, Lee H-I, Dean DR, Seefeldt LC, Hoffman BM (2005) J Am Chem Soc 127:12804–12805
Corbett MC, Hu Y, Fay AW, Ribbe MW, Hedman B, Hodgson KO (2006) Proc Natl Acad Sci USA 103:1238–1243
Acknowledgements
We want to thank all the former group members who contributed to some of the research reported here, particularly T. Lovell, R. Torres, F. Himo, J. Li, T. Liu, G.M. Ullmann, L. Hunsicker-Wang, J.-M. Mouesca, C.L. Fisher, R. Konecny, X.-G. Zhao, S. Sinnecker, and my collaborators D.A. Case, J.A. Fee, and D. Bashford. We thank V. Roberts, J.A. Fee, and V. Pelmenschikov for their critical reading and comments on the manuscript. This work was funded by NIH grants GM43278 and GM39914.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Noodleman, L., Han, WG. Structure, redox, pK a, spin. A golden tetrad for understanding metalloenzyme energetics and reaction pathways. J Biol Inorg Chem 11, 674–694 (2006). https://doi.org/10.1007/s00775-006-0136-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-006-0136-3