
Abstract
The axial ligand effect on reactivity of heme enzymes is explored by means of density functional theoretical calculations of the oxidation reactions of propene by a model compound I species of horseradish peroxidase (HRP). The results are assessed vis-à-vis those of cytochrome P450 compound I. It is shown that the two enzymatic species perform C=C epoxidation and C–H hydroxylation in a multistate reactivity scenario with FeIII and FeIV electromeric situations and two different spin states, doublet and quartet. However, while the HRP species preferentially keeps the iron in a low oxidation state (FeIII), the cytochrome P450 species prefers the higher oxidation state (FeIV). It is found that HRP compound I has somewhat lower barriers than those obtained by the cytochrome P450 species. Furthermore, in agreement with experimental observations and studies on model systems, HRP prefers C=C epoxidation, whereas cytochrome P450 prefers C–H hydroxylation. Thus, had the compound I species of HRP been by itself, it would have been an epoxidizing agent, and at least as reactive as cytochrome P450. In the enzyme, HRP is much less reactive than cytochrome P450, presumably because HRP reactivity is limited by the access of the substrate to compound I.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heme-type enzymes are abundant in nature and constitute roughly one third of all known metalloproteins [1]. Heme is the iron protoporphyrin IX prosthetic, FeIIIPor, shown in Scheme 1. This complex performs a great variety of chemical processes [1–6] depending on the protein, and as such, heme proteins have provided opportunities to probe how interactions between the protein and prosthetic group control function. An important functional difference in heme enzymes is the nature of the ligand LP, called the proximal ligand (Scheme 1). Two of the most common proximal ligands are thiolate of a cysteinate residue and the imidazole ligand of a histidine. Cytochromes P450 (P450s) have a thiolate ligand, while the great majority of peroxidases include a histidine proximal ligand [1]. The P450s are very versatile catalysts that activate dioxygen and insert a single oxygen atom into almost any imaginable organic compound [2]. For example, P450s can hydroxylate C–H bonds and epoxidate C=C bonds. Peroxidases, on the other hand, act mostly as electron sinks [1], converting H2O2 to water by performing single-electron oxidation of substrates. For instance, cytochrome c peroxidase oxidizes cytochrome c, itself an electron transport protein found in the mitochondria of eukaryotic organisms, and horseradish peroxidase (HRP) oxidizes phenolic compounds, leading to their coupling, etc. Interestingly, HRP is able to catalyze monooxygenation of thioethers, but it is a poor oxidant for C=C epoxidation and even poorer for C–H hydroxylation [7,8]. One of the more obvious factors that may differentiate P450 and HRP is the steric access of the substrate to the active species [7, 9]. However, much less established are the intrinsic differences between the active species of the two enzymes, owing to the different proximal ligands. For example, the use of histidine-based microperoxidases [10] suggests that the histidine (imidazole) ligand inhibits the reactivity relative to P450, whereas working with model compounds leads to much less decisive conclusions regarding the preferred axial ligand for monooxygenation. Thus, the main goal of the present paper is to benchmark the intrinsic reactivity differences between the active species of heme enzymes that are based on thiolate (P450-like) and those based on histidine (HRP-like). Are there any ligand influences on the relative reactivity and regiochemistry during monooxygenation?

The active species of HRP is the high-valent iron-oxo porphyrin called compound I (Cpd I). The same species is believed to be active in P450 [1]. Scheme 1 shows the X-ray structures of the Cpd I species of HRP and P450, taken from the protein data bank [11–13]. The amino acids are labeled in accordance with the labeling of the X-ray structures.
Since, histidine is a neutral ligand, while cysteinate is an anionic ligand, the electrostatic effects induced by the ligands on the catalytic centers are very different for HRP and P450. Thus, it has been shown that cysteinate pushes electrons, whereas histidine pulls electrons [3, 14, 15]. One of the results is that in HRP-related systems the iron atom is located more in the plane of the heme group, whereas in P450 enzymes it rises above the plane [14]. However, the local environments of the ligands are also different in the two enzymes [1, 14]. The histidine ligand in HRP is located at the C-terminal of the proximal helix which exerts a negative environment, whereas the cysteinate ligand in P450 resides at the electropositive N-terminus of the helix. A decrease of the negative charge on the ligand has been argued to increase the Fe(3+)/Fe(2+) reduction potential [14]. Another environmental difference between the cysteinate and the histidine axial ligands is that histidine donates a hydrogen bond (to Asp247, Scheme 1), while cysteinate accepts amidic hydrogen bonds [14]. Mutation of the axial ligand in myoglobin from histidine to cysteine leads to a mutant enzyme that possesses nearly identical spectroscopic features as P450, and its reactivity patterns suggest that it forms Cpd I during oxidative processes [15]. This suggests that in dealing with the intrinsic reactivities of the two species, one might be allowed to use similar representations of the “polarity” of the environments.
The intrinsic effect of the axial ligand has been tested using synthetic oxo-iron porphyrin catalysts. Cyclooctene/cyclooctane oxidation by wild-type P450 gives a hydroxylation/epoxidation product ratio of 4.04. However, synthetic thiolate ligated oxo-iron porphyrin systems change the regioselectivity in favor of epoxidation (product ratio 0.66), whereas synthetic imidazole ligated catalysts yield dominant epoxidation [16]. In the case of O-demethylation of alkyl aryl esters, a catalyst with a thiolate axial ligand even changes the reaction mechanism compared with a catalyst having chloride or imidazole axial ligands [17]. Studies with various axial ligands on synthetic oxo-iron porphyrin systems showed that some catalysts react via FeIII intermediates, while others react via FeIV intermediates and that therefore, the reaction mechanism may vary with the nature of the axial ligand [18–20]. Moreover, the axial ligand may enhance the oxidative power of Cpd I towards one-electron reduction and formation of the one-electron reduced species, compound II. In this sense, the electron release property of thiolate makes Cpd I of P450 a much less powerful electron acceptor than HRP Cpd I; the electron affinities are computed to be 3.06 versus 6.41 eV, respectively [21].
Although many studies have addressed the electronic properties of P450 and HRP Cpd I species [22–30], at this point of time no comparative reactivity studies have been performed theoretically. The present paper describes density functional theoretical calculations, which gauge the intrinsic effects of the proximal ligands (thiolate and imidazole) on the hydroxylation and epoxidation reactions of Cpd I models of P450 and HRP, and assess the effect of the polar environment on the relative barriers of the two reactions. As a substrate, which contains the C=C and C–H functionalities, we use propene, which was investigated before in combination with a Cpd I model of P450, and was found to exhibit tantalizing regioselectivity crossovers brought about by the polarity of the environment and its hydrogen-bonding capability [31, 32]. These latter features of the reaction will be addressed here too, for HRP Cpd I, and a comparison will be made between the two Cpd I species.
Methods
There is a question to what extent the hydrogen bonding between the imidazole and the neighboring aspartate affects the “imidazolate character” of the proximal ligand in HRP Cpd I [25, 26, 29]. Using imidazolate leads to significant spin density transfer from the porphyrin to the proximal ligand, whereas the use of imidazole as the ligand leads to negligible spin density transfer [26]. Our previous study of HRP Cpd I [21] which included imidazole and aspartate as well as the side chain of phenylanaline (Scheme 1) showed that there is indeed a hydrogen bond between imidazole and aspartate, but the resulting imidazolate character and the extent of the spin density on the ligand are small; when the effect of a dielectric medium is included, most of the spin density resides on the porphyrin. Following this study [21] the HRP Cpd I species was modeled as an oxo-iron porphine with an imidazole axial ligand. P450 Cpd I was modeled using thiolate (SH−) as axial ligand; previous calculations showed that the gas-phase results for this ligand are closer than other representations of the cysteinate ligand to the full quantum mechanical/molecular mechanical results [33, 34].
All the calculations followed commonly used procedures [31, 32] that are briefly summarized here. We used the unrestricted hybrid density functional method UB3LYP [35–38] in combination with a double-ζ-quality basis set (LACVP) [39, 40] on iron and a Pople-type 6-31G basis set on the remaining atoms, hence LACVP(iron)/6-31G(rest) [41]. The effect of increasing the size of the basis set was tested using a single-point calculation with the triple-ζ polarized basis set, which is augmented by diffuse functions, LACV3P+*(iron) 6-311+G*(rest) [39, 40]. Essentially the same trends were found for the two basis sets. The geometries were fully optimized using the Jaguar 4.2 program package [42] followed by an analytic frequency in Gaussian 98, using the same basis set LACVP(iron)/6-31G(rest) [43].
The connections between intermediates and products, and between intermediates and reactants, were verified by running extensive geometry scans between the critical points. In these scans one degree of freedom was used as a reaction coordinate, whereas all other degrees of freedom were fully optimized. Subsequently, the tops of the scans were refined by full optimizations in a transition-state search. All transition states and intermediates were verified by frequency calculations. The effect of the environment was tested with the polarized continuum model as implemented in Jaguar 4.2 using a dielectric constant of ɛ=5.7 and a probe radius of 2.72 Å. The many pieces of data are summarized in the “Electronic supplementary material”.
Results
HRP Cpd I versus P450 Cpd I
Prior to discussing the reactivity pattern of HRP versus P450 enzymes, let us start with a brief summary of the important differences and comparisons of the two active species [21]. Scheme 2 shows the high-lying occupied and low-lying virtual orbitals of HRP Cpd I and P450 Cpd I. Both species have a set of five metal-type d orbitals, which from bottom to top are labeled as δ, \( \pi _{xz}^ * \), \( \pi _{yz}^ * \), \( \sigma ^{ * }_{{z^{2} }} \) and \(\sigma _{xy}^ * \). The δ orbital is nonbonding and lies in the plane of the porphyrin ring. Two orbitals \((\pi _{xz}^ * {\text{ and }}\pi _{yz}^ * ) \) represent the antibonding combinations between the metal 3dxz,yz iron orbitals with the 2px,y orbitals on oxygen. Two virtual \(\sigma ^ *\) orbitals with antibonding interactions of the 3d xy orbital with the nitrogen atoms of the porphyrin ring \( (\sigma _{xy}^ * ) \) and an antibonding interaction along the O–Fe–ligand axis (\( \sigma ^{ * }_{{z^{2} }} \)) complete the set of five metal orbitals. Save minor mixings owing to the low symmetry of the species, the d-block orbitals are rather similar for the two iron-oxo models (“Electronic supplementary material”, Fig. S6). Another high-lying orbital is a porphyrin-type a2u orbital, which is singly occupied in the two species. But, in P450, this orbital mixes strongly with the thiolate σS sulfur orbital in antibonding fashion, whereas in HRP it is virtually porphyrin centered [21].

High-lying occupied and low-lying virtual orbitals of HRP Cpd I (left) and P450 Cpd I (right). In both cases we show the occupancy and spin situation for the quartet A2u state.
The thiolate ligand is known to exert, on the rest of the molecule, a so-called “push effect” that is associated with the electron-releasing property of the ligand and its strong binding to iron [3]. Indeed, all the orbitals of HRP Cpd I are significantly lower in energy than the corresponding ones in P450 Cpd I, and this is especially so for the a2u orbitals. The relative energy levels of the orbitals for the two species reflect the “push effect”, and are mainly caused by the different charges of the axial ligands: thiolate is negative, while imidazole is neutral. Bonding and antibonding features play a secondary role, and are nonnegligible in the case of a2u and \( \sigma ^{ * }_{{z^{2} }} \) [44]. Thus, the destabilization of the a2u orbital, due to the negative charge and the antibonding interaction of the thiolate ligand, is responsible in part for the smaller electron affinity, 3.06 eV, of the P450 Cpd I species, compared with 6.41 eV, for HRP Cpd I [21]. Interestingly, the a2u orbital energy difference for the two species, approximately 3.4 eV, is very close to the electron affinity difference (3.35 eV).
Despite these differences, both P450 Cpd I and HRP Cpd I have ground states with \( \pi *^{1}_{{xz}} \,\pi *^{1}_{{yz}} \,a_{{{\text{2}}u}} ^{1} \) occupations that lead to doublet and quartet spin states, which are virtually degenerate; hereafter these states are designated as 4,2A2u. The state ordering is dependent on the model investigated as well as on the environment [21, 29, 30, 33]. However, in any model, the two states are virtually degenerate.
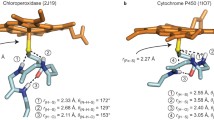
Optimized geometries of the lowest-lying quartet and doublet spin states (4,2A2u) are shown in Fig. 1. Since, the thiolate ligand is a better iron binder compared with imidazole [30, 44], it will exert a trans effect (bond lengthening) on the oxo ligand or any ligand trans to thiolate. Indeed, the iron-oxo bond is longer: 1.651 Å in the 4 A2u state of P450 Cpd I compared with 1.621 Å in the same state of HRP Cpd I. The trans effect of thiolate is reflected also in the displacement of the iron atom with respect to the plane of the porphyrin ring (Δ), which in the P450 optimized geometry is larger than in the corresponding HRP species. In fact, the “push” and trans effects of thiolate are synonymous, both originating from the negative charge and strong binding of this ligand to iron (good orbital mixing) [44], compared with imidazole.

Optimized geometries of the 4,2A2u ground states of horseradish peroxidase (HRP) (left) and cytochrome P450 (P450) (right) compound I (Cpd I) species. All bond lengths are in angstroms; Δ is the displacement of the iron atom with respect to the plane of the porphyrin ring and \(\bar r_{{\rm{FeN}}} \) is the average value of the four Fe–Npyrrole distances.
Propene epoxidation by HRP Cpd I
Figure 2 displays the potential energy profile as well as optimized geometries of the transition states and intermediates which were found in the epoxidation of propene by the HRP model Cpd I; the energy values in square brackets correspond to the larger basis set LACV3P+*. The initial phase of the reaction involves the C=C bond activation, followed by a ring-closure phase to give the epoxide complex. Four different transition states were located for the bond-activation phase: two with the metal in oxidation state FeIV and two with the metal in oxidation state FeIII. The two FeIV-type transition states (4,2TS1-IV) are close in energy, within 0.6–1.1 kcal/mol. This is in good agreement with our previous results with P450 Cpd I. By contrast, the lowest-energy transition state is 2TS1-III, which involves FeIII and is 5.33 [8.47] kcal mol−1 above the isolated reactants. In the P450 case, this latter transition state is high in energy [31, 32].

Potential energy profile for the epoxidation of propene (P) using 4,2Cpd I (HRP). All energies are relative to 4Cpd I+propene, are in kilocalories per mol and contain zero-point-energy (ZPE) corrections. The energy values in square brackets correspond to single-point energy calculations with LACV3P+*, including the ZPE correction from LACVP. Bond lengths are in angstroms. The asterisk near 4TS2-III means that the energy value of this structure was estimated from scans, and involves only LACVP data.
After passing the initial bond activation barriers, the high-spin (HS) pathways generate two electromeric intermediates (42-IV and 42-III) that have ring-closure barriers, which are 0.58 (via 4TS2-IV) and 2.87 kcal mol−1 (via 4TS2-III) above the respective intermediates. The FeIII-optimized complexes show longer Fe–O bond distances owing to an additional electron (compared with the FeIV electromer) in the \( \pi _{xz}^ * \) orbital, which has antibonding character in the Fe–O linkage. By contrast, on the low-spin (LS) surfaces we did not manage to locate real intermediates. After passing the bond activation step on the LS surfaces, the species directly collapsed to the product complexes in an effectively concerted fashion. Therefore, the LS pathway will be effectively concerted (albeit nonsynchronous), while the HS pathway that proceeds via 4TS2-III will exhibit a stepwise mechanism with a real intermediate having finite lifetimes. This will become more pronounced in a polarizing environment, as shown recently for the P450cam case where the protein increased the rebound barrier for the Fe(III) state [45]. This may result in stereochemical scrambling or rearrangement reactions on the HS surface.
As seen from the energy values in brackets, the trends in this scenario remain basically invariant with the larger basis set;hence we shall focus in the rest of the paper on the LACVP results.
Propene hydroxylation by HRP Cpd I
Figure 3 shows the potential energy profile for the allylic hydroxylation of the methyl group of propene by HRP Cpd I. Similarly to the epoxidation mechanism, here too a multistate scenario is clearly visible. The initial step involves hydrogen abstraction and leads to an intermediate of the hydroxo iron complex with a nearby allyl radical. This allyl radical, thereafter, rebounds onto the iron hydroxo to form the propenol product complex. As can be seen from Fig. 3, the lowest-lying pathways for each spin manifold are with the iron in oxidation state FeIII, while the FeIV states are 2 kcal mol−1 or more higher in energy. In the P450 case, the FeIII transition states are very high in energy [32]. As in the case of epoxidation, here too the two HS intermediates encounter barriers for the formation of the alcohol complexes. By contrast to the epoxidation pathways, the LS pathways also pass via radical species before collapsing to the LS product complex. However, these LS species are shoulders and not true intermediates, they have no rebound barrier, so their lifetime will be limited.

Potential energy profile for the hydroxylation of propene using 4,2Cpd I (HRP). All energies are relative to 4Cpd I + propene, are in kilocalories per mole and contain ZPE corrections. The energy values in square brackets correspond to the results with the LACV3P+* basis set with ZPE inclusion from LACVP. Bond lengths are in angstroms. The asterisk near 4TS4-III means that the energy value of this structure was estimated from scans, and involves only LACVP data.
Environmental effects on barrier heights
To examine the effect of a polarizing environment, the lowest transition state species were recalculated, using a medium with a dielectric constant ɛ=5.7. Figure 4 shows these results for the reactions with HRP Cpd I, compared with those of P450 Cpd I. It is seen that a dielectric medium stabilizes the hydroxylation transition states more than the corresponding epoxidation species. This however is insufficient to make the hydroxylation pathways favorable over epoxidation; the pathway via 2TS1-III is still the lowest-lying. Using the LACV3P+* data leads basically to the same trend (“Electronic supplementary material”, Table S7). Thus, even though the differences are small, they seem to persist. We may therefore conclude that HRP Cpd I would intrinsically prefer epoxidation to hydroxylation. In the case of P450 (Fig. 4, right), a dielectric constant of ɛ=5.7 creates a preference for hydroxylation over epoxidation, by 1.4 kcal mol−1, and this further increases to 3.0 kcal mol−1 when the sulfur is hydrogen-bonded by NH...S hydrogen bonds. Therefore, a polar environment as applied by the protein environment will give preferential epoxidation in HRP enzymes and dominant hydroxylation in P450 enzymes, in agreement with experimental observations on model systems [16]. This, of course, is an intrinsic property of the corresponding Cpd I, and it does not consider factors such as the access of the substrate to Cpd I and the tightness of the protein pocket.

Relative energies of the lowest-lying 4,2TS1 (epoxidation) and 4,2TS3 (hydroxylation) transition states in the oxidation of propene by HRP Cpd I (left) and by P450 Cpd I (right) under different external perturbations; ɛ is a dielectric constant. The relative energies include the ZPE correction in the gas-phase energies.
Discussion
The goal of this paper was to gauge the intrinsic reactivity of HRP and P450, with an aim of understanding whether the sluggish monooxygenation reactivity of HRP [7– 9] is an intrinsic property of the system, or whether is it due to the constraints exerted by the protein pocket. To facilitate this discussion, we collected the barriers for epoxidation and hydroxylation for HRP and P450 Cpd I species in Table 1. The effect of a dielectric medium can be gleaned from Fig 4.
Intrinsic differences between P450 and HRP
The rate-determining step proceeds via 2,4TS1 (epoxidation) and 2,4TS3 (hydroxylation). Comparing the lowest-energy species, we see that the corresponding barriers are 2–5 kcal mol−1 lower in HRP systems than in P450 systems. The group spin densities and key bond distances of the lowest-lying transition states 2TS1 and 2TS3 are displayed in Fig. 5. As can be seen, these LS transition states for the reaction of HRP involve less O–C and O–H bond making and less C=C and C-H bond breaking compared with the P450 transition states, and in the case of the epoxidation mechanism also a smaller amount of spin density development on the propene compared with the P450 case. As such, the HRP species are “early”, while the P450 ones are more “advanced” transition states. Based on the Hammond postulate, the HRP reactions should have smaller barriers than the P450 reactions. The calculated barrier differences are in accord with this prediction. Therefore, with due caution, we may conclude that, intrinsically, the HRP Cpd I species should be at least as reactive as the corresponding P450 species. The experimental data [7–9] show that the monooxygenation reactivity of HRP is sluggish (especially with respect to C-H hydroxylation), and our results indicate that this sluggishness is not intrinsic; this may well be a result of the topology of the protein pocket that restricts substrate access to Cpd I.

Group spin densities and key bond distances of the lowest-energy transition states of the TS1 and TS3 varieties for the reactions of HRP Cpd I (left) and P450 Cpd I (right)
Another interesting feature is the regiochemistry of propene oxidation. HRP Cpd I prefers C=C epoxidation over C–H hydroxylation, and this preference carries over to a polarizing environment (Fig. 4). By contrast, P450 Cpd I exhibits in the gas phase a slight intrinsic preference for C=C epoxidation. This latter trend is completely reversed when polarization and hydrogen-bonding effects are taken into account. This is in agreement with experimental observations regarding the activity of model systems [16–19]. The fact that the imidazole group in HRP is located in a negatively charged region, whereas the cysteinate of P450 is located in a positively charged region [14], may further enhance the regioselectivity differences as shown by us recently using applied electric fields to modulate the regiochemistry of P450 oxidation [46].
Finally the product complexes also exhibit a difference in the spin quantum numbers of the ground states. In propene oxidation by P450, this state is generally the LS doublet state [32]. This is also true for other reactions, as well as for the resting state of P450 [31, 32, 46–50]. In HRP, by contrast, we find here 43 below 23 and 45 below 25. The energy differences increase upon improvement of the basis set (Figs. 2, 3, “Electronic supplementary material”, Table S7, for LACV3P+* data), so there is a trend here to be reckoned with.
The energy difference between HS and LS complexes should reflect as usual a balance of a few factors: (1) the orbital energy gap \( (\pi _{xz}^ * {\text{ and }}\sigma _{z^2 }^ * ) \) prefers the doublet state, (2) the exchange interaction of the identical-spin electrons prefers the quartet state, and (3) the different geometries of the complexes in the two spin states. Our search showed that the orbital energy gaps for the product complexes are virtually the same, and the corresponding geometries do not give much of a clue either. What seems to matter most is that the \( \sigma ^{ * }_{{z^{2} }} \) orbitals of the P450 product complexes are more delocalized, involving substantial contribution from the sulfur (see the orbitals in Fig. S7 in the “Electronic supplementary material”). On the other hand, in the HRP product complexes, these orbitals are more localized on the iron. Consequently, the exchange interaction energy in the HRP HS complex will be more stabilizing than in the corresponding P450 complex. As such, once again, the better binding capability of the thiolate ligand affects the relative stability of the spin states. This inversion in the relative stability of the two spin states of the product complex creates an additional spin crossover junction, thus further enhancing the entangled reactivity of the two spin states, and would affect the restart of the catalytic cycle of the enzyme or of a Cpd I species under turnover conditions.
Conclusion
The results of this study show intrinsic differences between the reactivity patterns of the Cpd I species of P450 and HRP. The major findings are (1) Cpd I of HRP is intrinsically more or at least as reactive than P450 Cpd I, (2) HRP prefers epoxidation, while P450 prefers C–H hydroxylation, and (3) the most favorable pathway in HRP is the LS C=C epoxidation, which proceeds to the epoxide complex without an intermediate; the HS pathways are significantly higher in energy. As such, one would expect to find fewer side products during epoxidation, e.g., suicidal complexes and aldehyde complexes [2–4, 51]. Our results are in good general accord with experimental trends based on model systems [16, 17]. The higher intrinsic reactivity of the HRP species is, however, not in accord with the opposite trend suggested from studies of microperoxidases [10].
These differences identified in our study trace to the charges of the axial ligands (imidazole versus thiolate) and the extents of mixing of their orbitals into the d orbitals of the iron and the a2u orbital of the porphyrin [44]. Owing to the negative charge of the thiolate and the better mixing of its orbitals with those of the heme, this ligand is a better iron binder compared with imidazole [30, 44]. Consequently, the d-block and a2u orbitals are considerably higher in energy for the thiolate complexes, and this is the root cause behind most of the intrinsic differences between the P450 and HRP species. In the language of the inorganic chemistry community, these orbital patterns cause a pronounced “trans effect” in the case of thiolate, and, in the language of bioinorganic chemistry community, this translates into the “push effect” of this ligand [3]. While our study did not consider the scenario of imidazolate as a proximal ligand [26], our analysis nevertheless suggests that imidazolate would have made HRP Cpd I more similar to P450 Cpd I.
The computational findings further suggest that, in principle, one can design appropriate HRP mutants that will be selective epoxidation catalysts, which also perform stereospecific reactions. Some HRP mutants [7–9] and microperoxidases [10] that are mildly potent monooxygenating species are known and may serve as guides for future theoretical studies focused on the design of new potent mutants.
References
Poulos TL (2000) In: Kadish KM, Smith KM, Guilard R (eds) The porphyrin handbook, Volume 4, Chapter 32, pp 189–218
Cytochrome P450: structure, mechanism and biochemistry, 2nd edn; Ortiz de Montellano PR (Ed) Plenum Press, New York (1995)
Sono M, Roach MP, Coulter ED, Dawson JH (1996) Chem Rev 96:2841–2888
Guengerich FP (2001) Chem Res Toxicol 14:611–650
Ortiz de Montellano PR, De Voss JJ (2002) Nat Prod Rep 19:477–494
Groves JT (2003) Proc Natl Acad Sci U S A 100:3569–3574
Ozaki SI, Ortiz de Montellano PR (1995) J Am Chem Soc 117:7056–7064
Ortiz de Montellano PR (1992) Annu Rev Pharmacol Toxicol 32:89–107 (For oxygenase activity of HRP)
Ator MA, Ortiz de Montellano PR (1987) J Biol Chem 262:1542–1551 (Phe41 restricts access of the substrate to HRP Cpd I)
Rietjens IMCM, Osman AM, Veeger C, Zakharieva O, Antony J, Grodzicki M, Trautwein AX (1996) J Biol Inorg Chem 1:372–376
Berglund GI, Carlsson GH, Smith AT, Szöke H, Henriksen A, Hajdu J (2002) Nature 417:463–468
Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG (2000) Science 287:1615–1622
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) Nucleic Acids Res 28:235–242
Poulos TL (1996) J Bioinorg Chem 1:356–359
Matsui T, Nagano S, Ishimori K, Watanabe Y, Morishima I (1996) Biochemistry 35:13118–13124
Ohno T, Suzuki N, Dokoh T, Urano Y, Kikuchi K, Hirobe M, Higuchi T, Nagano T (2000) J Inorg Biochem 82:123–125
UranoY, Higuchi T, Hirobe M, Nagano T (1997) J Am Chem Soc 119:12008–12009
Gross Z, Nimri S (1994) Inorg Chem 33:1731–1732
Gross Z (1996) J Bioinorg Chem 1:368–371
Nam W, Lim MH, Oh SY, Lee JH, Lee HJ, Woo SK, Kim C, Shin W (2000) Angew Chem Int Ed 39:3646–3649
de Visser SP, Shaik S, Sharma PK, Kumar D, Thiel W (2003) J Am Chem Soc 125:15779–15788
Loew GH, Kert CJ, Hjelmeland LM, Kirchner RF (1977) J Am Chem Soc 99:3534–3536
Du P, Loew GH (1995) Biophys J 68:69–80
Kuramochi H, Noodleman L, Case DA (1997) J Am Chem Soc 119:11442–11451
Deeth RJ (1999) J Am Chem Soc 121:6074–6075
Green MT (2000) J Am Chem Soc 122:9495–9499
Wirstam M, Blomberg MRA, Siegbahn PEM (1999) J Am Chem Soc 121:10178–10185
Loew GH, Harris DL (2000) Chem Rev 100:407–420 For reviews on Cpd I of P450
Harris DL (2001) Curr Opin Chem Biol 5:724–735
Rydberg P, Sigfridsson E, Ryde U (2004) J Biol Inorg Chem 9:203–223
De Visser SP, Ogliaro F, Sharma PK, Shaik S (2002) Angew Chem Int Ed 41:1947–1951
De Visser SP, Ogliaro F, Sharma PK, Shaik S (2002) J Am Chem Soc 124:11809–11826
Ogliaro F, de Visser SP, Cohen S, Kaneti J, Shaik S (2001) Chem Bio Chem 2:848–851
Schöneboom JC, Lin H, Reuter N, Thiel W, Cohen S, Ogliaro F, Shaik S (2002) J Am Chem Soc 124:8142–8151
Becke AD (1992) J Chem Phys 96:2155–2160
Becke AD (1992) J Chem Phys 97:9173–9177
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Hay JP, Wadt WR (1985) J Chem Phys 82:299–308
Friesner RA, Murphy RB, Beachy MD, Ringlanda MN, Pollard WT, Dunietz BD, Cao YX (1999) J Phys Chem A 103:1913–1928
Hehre WJ, Ditchfield R, Pople JA (1972) J Chem Phys 56:2257–2261
Jaguar 4.2; Schrödinger, Inc.: Portland, OR, 2000
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA,Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY Nanayakkara A, Gonzalez C, Challacombe M, Gill PMW, Johnson BG, Chen W, Wong MW, Andres JL, Head-Gordon M,Replogle ES, Pople JA, Gaussian, Inc.: Pittsburgh, PA, 1998
Ogliaro F, de Visser SP, Shaik S (2002) J Inorg Biochem 91:554–567
Schöneboom JC, Cohen S, Lin H, Shaik S, Thiel W (2004) J Am Chem Soc 126:4017–4034
Shaik S, de Visser SP, Kumar D (2004) J Am Chem Soc 126:11746–11749
De Visser SP, Shaik S (2003) J Am Chem Soc 125:7413–7424
Kumar D, de Visser SP, Shaik S (2003) J Am Chem Soc 125:13024–13025
Kumar D, de Visser SP, Sharma PK, Cohen S, Shaik S (2004) J Am Chem Soc 126:1907–1920
Schöneboom JC, Thiel W (2004) J Phys Chem B 108:7468–7478
de Visser SP, Kumar D, Shaik S (2004) J Inorg Biochem 98:1183–1193
Acknowledgement
A grant (to S.S.) by the Israel Science Foundation is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
The paper is dedicated to D. K. Bohme on the occasion of his forthcoming 65th birthday.
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Kumar, D., de Visser, S.P., Sharma, P.K. et al. The intrinsic axial ligand effect on propene oxidation by horseradish peroxidase versus cytochrome P450 enzymes. J Biol Inorg Chem 10, 181–189 (2005). https://doi.org/10.1007/s00775-004-0622-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-004-0622-4