
Abstract
Electron nuclear double resonance (ENDOR) and hyperfine sublevel correlation spectroscopy (HYSCORE) are applied to study the active site of catalytic [NiFe]-hydrogenase from Desulfovibrio vulgaris Miyazaki F in the reduced Ni-C state. These techniques offer a powerful tool for detecting nearby magnetic nuclei, including a metal-bound substrate hydrogen, and for mapping the spin density distribution of the unpaired electron at the active site. The observed hyperfine couplings are assigned via comparison with structural data from X-ray crystallography and knowledge of the complete g-tensor in the Ni-C state (Foerster et al. (2003) J Am Chem Soc 125:83–93). This is found to be in good agreement with density functional theory calculations. The two most strongly coupled protons (aiso=13.7, 11.8 MHz) are assigned to the β-CH2 protons of the nickel-coordinating cysteine 549, and a third proton (aiso=8.9 MHz) is assigned to a β-CH2 proton of cysteine 546. Using D2O exchange experiments, the presence of a hydride in the bridging position between the nickel and iron—recently been detected for a regulatory hydrogenase (Brecht et al. (2003) J Am Chem Soc 125:13075–13083)—is experimentally confirmed for the first time for catalytic hydrogenases. The hydride exhibits a small isotropic hyperfine coupling constant (aiso=−3.5 MHz) since it is bound to Ni in a direction perpendicular to the z-axis of the Ni \( {\left( {3d_{{z^{2} }} } \right)} \) orbital. Nitrogen signals that belong to the nitrogen Nε of His-88 have been identified. This residue forms a hydrogen bond with the spin-carrying Ni-coordinated sulfur of Cys-549. Comparison with other hydrogenases reveals that the active site is essentially the same in all proteins, including a regulatory hydrogenase.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hydrogenases comprise a fundamental group of bacterial enzymes that catalyze the reversible oxidation of molecular hydrogen to protons, and play a vital role in the anaerobic metabolisms of a wide variety of microorganisms [1, 2]. Three phylogenetically distinct classes of hydrogenases have been discovered so far, that differ in terms of their metal content [3]. Among those, [NiFe]-hydrogenases represent the largest class. Many organisms contain two or more different [NiFe]-hydrogenases situated in different cell compartments. Their function (hydrogen uptake or production) is associated with their respective locations in the cell. The diversity of the hydrogenases in these bacteria demonstrates the important role of hydrogen in their metabolism. Moreover, regulatory [NiFe]-hydrogenases are also present, which ensure a rapid and efficient response to variations in energetic needs under changing growth conditions.
During catalysis, hydrogenase cycles through various redox states, several of which are paramagnetic. The oxidized enzyme displays two different EPR signals, termed Ni-A (“unready”) and Ni-B (“ready”) [4, 5]. Under reductive conditions another characteristic EPR signal appears, named Ni-C (“active”). This signal is associated with the catalytically active state since it is also observed during hydrogenase catalysis in vivo [6]. The reduced form of hydrogenase is capable to catalyze proton–deuterium or proton–tritium exchange in the absence of electron donors or acceptors, according to the reaction H2 + D2O → HD + HDO. This finding indicates the direct accessibility of the active site in the Ni-C state to molecular hydrogen. Furthermore, it suggests that molecular hydrogen is heterolytically cleaved, producing a proton and a hydride and the subsequent formation of an enzyme-hydride intermediate [7–9].
The [NiFe]-hydrogenase of D. vulgaris Miyazaki F consists of a large and a small subunit. The crystal structure of the enzyme has been solved to 1.8 Å resolution [10]. Three [FeS] clusters are present in the small subunit and the active site is buried deeply in the large subunit. The active site is depicted schematically in Fig. 1 and consists of iron and nickel that are bridged by two cysteines. The nickel is further ligated by two terminal cysteines and the iron has three diatomic inorganic ligands. Although the conformation of the active site is found to be conserved for many [NiFe]-hydrogenases [10–14], disagreement between the crystal structures exists with respect to the nature of the diatomic ligands to iron. In D. vulgaris Miyazaki F they are assigned to one SO and two CN ligands [10], whereas in all other crystal structures one CO and two CNs are found [11–14]. Recent infrared absorption measurements have, however, unequivocally established that the [NiFe]-hydrogenase of D. vulgaris Miyazaki F also has one CO and two CN− ligands (C. Fichtner et al., Max-Planck-Institut für Bioanorganische Chemie, unpublished data, 2004).

In the crystal structures of the as-isolated, oxidized states Ni-A and Ni-B, additional electron density is found between Ni and Fe, which stems from a third bridging ligand. This ligand is most likely an oxygen ligand [11, 15–17]. In the active state, called Ni-C, which is two electrons more reduced than Ni-A and Ni-B, this electron density is absent [18, 19]. A large 1H hyperfine interaction has been found in electron nuclear double resonance (ENDOR) experiments [20, 21], from which a hydride bridge for the reduced state was postulated. Recently, the complete hyperfine tensor of the hydride bridge was determined from hyperfine sublevel correlation (HYSCORE) spectroscopy of the regulatory hydrogenase of Ralstonia eutropha in the Ni-C state [22].
In order to understand the reaction mechanism of the [NiFe]-hydrogenase, details about the electronic structure of the active site are required. Recently, we determined the orientation of the g-tensor of the Ni-C state for D. vulgaris Miyazaki F hydrogenase by single crystal EPR spectroscopy [23]. The principal axis that corresponds to the smallest g value (g z or g3) was found to lie almost parallel to the bond direction of Ni and the sulfur of Cys-549 (one of the bridging cysteines), indicating a Ni(III) \( d_{{z^{2} }} \) ground state with the C∞ symmetry axis of the \( d_{{z^{2} }} \) orbital parallel to this bond direction.
Spin density distributions for the paramagnetic states of [NiFe]-hydrogenases have been calculated by several groups using density functional theory (DFT) [16, 24, 25]. Although disagreement exists over the interpretations of certain aspects of the calculations, the overall spin density distribution in all calculations is compatible with the direction of the \( d_{{z^{2} }} \) C∞ symmetry axis, as was found in the single crystal EPR study [23].
In parallel to our EPR study, the orientation of the g-tensor of the Ni-C state of two other enzymes, the [NiFe]-hydrogenase of D. gigas and the [NiFeSe]-hydrogenase of D. baculatum, which has a selenocysteine as a ligand to Ni, was indirectly determined from orientation-selected 1H ENDOR spectroscopy on frozen solutions [26]. In this study, a comparison of the 1H ENDOR signals from these two enzymes was performed and it was found that the ENDOR signals from both enzymes were very similar, which led to the conclusion that the active sites of the two proteins are essentially identical. Nevertheless, the g-tensor elucidated from these data differs significantly from the one for D. vulgaris Miyazaki F and from the ones calculated by DFT on a [NiFe]-hydrogenase model system. The g z principal axis (called g3 in reference [23]) is found to make an angle of 37° with the Ni-S (Cys 549) (Cys-533 for D. gigas) direction [26]. An angle of only 5° was found for Ni-C in D. vulgaris Miyazaki F [23]. The orientations of the g x and g y axes differ by more than 50° in these studies.
In order to obtain more detailed information about the electronic structure of the Ni-C state and to solve this discrepancy we performed orientation-selected 1H ENDOR spectroscopy and HYSCORE spectroscopy on D. vulgaris Miyazaki F [NiFe]-hydrogenase. Proton hyperfine couplings were deduced for three non-exchangeable protons from ENDOR. HYSCORE spectroscopy showed that the hydride bridge, as found for the regulatory hydrogenase of R. eutropha, is also present in the standard hydrogenases. The results are compared to those obtained for D. gigas hydrogenase and those derived from DFT calculations to obtain a consistent picture of the electronic structure of the reduced Ni-C state.
Materials and methods
Sample preparation
The [NiFe]-hydrogenase from D. vulgaris Miyazaki F was isolated and purified as described previously [27]. Tris/HCl buffer for the solvent exchange was prepared by thorough lyophylization of 1 ml portions of the respective buffer solution in H2O and redilution in the solvent. To achieve 25 mM Tris/DCl pDtrue=7.4, a dried solution of Tris/HCl at pH 7.1 was used [28, 29]. Solvent exchange to D2O (99.9 atom %) was achieved by concentrating the enzyme solution with a microconcentrator to about 1/20 of the initial volume and re-diluting it six times at 5 °C with the respective buffer solution in D2O.
Desulfovibrio vulgaris hydrogenase samples were equilibrated in the EPR tubes in a pure hydrogen gas atmosphere for 2–3 h, yielding the Ni-C state. Samples with an increased fraction of the “unsplit” Ni-C form were generated by purging the reduced enzyme for 15–30 min with argon as described in [30]. The fraction of unsplit Ni-C was estimated to be 50–70% according to the relative intensities of the EPR signals of the split and unsplit form at g y and g x . Reduction of the enzyme in D2O was undertaken in 100% D2 gas atmosphere following the procedure described above.
EPR, ENDOR, and HYSCORE spectroscopy
EPR spectra were measured on a Bruker ESP 300E spectrometer equipped with a Bruker 4105 DR resonator. The cw-ENDOR measurements were performed on the same type of spectrometer using a home-built TM110 ENDOR resonator especially designed for low-temperature experiments on transition metal systems [31]. A 16-turn ENDOR coil was used for the 1H ENDOR experiments. Furthermore, a radio frequency (RF) signal generator (Rhode-Schwarz, SMT 02), and a 200 W RF power amplifier (ENI A3200 L, 0.25–150 MHz) were employed. Both set-ups were equipped with an Oxford helium cryostat ESR 910 (1.8–300 K), the field was calibrated with a Bruker ER035 NMR-Teslameter and the microwave frequency was measured with a Hewlett-Packard 5352B frequency counter.
Pulse EPR and HYSCORE measurements were performed with a Bruker ESP 380E spectrometer in a Bruker dielectric ring resonator (ESP380-1052 DLQ-H) equipped with an Oxford helium cryostat CF 935. Temperatures below 4.2 K were obtained by decreasing the pressure in the cryostat with an additional pump. In the 2D-HYSCORE experiment, the pulse sequence of π/2-τ-π/2-t1-π-t2-π/2-τ-echo [32, 33] was applied with π/2 and π pulses of 16 and 32 ns, respectively. A four-step phase cycle suggested by Gemperle et al. [34] was used to eliminate contributions from unwanted echoes from the echo envelopes. The baseline of the 2D modulation pattern was subtracted using a second-order polynomial background function. Subsequently, the time traces were multiplied with a Hamming function and zero-filled to 512×512 points. The data were then Fourier-transformed to obtain magnitude contour spectra.
Simulation procedures
The EPR powder spectra were simulated using an in-house simulation program [35]. Orientation-selected ENDOR spectra were simulated using the program SPLEEN [36]. This program computes ENDOR transition frequencies and intensities for an arbitrary number of paramagnetic nuclei, performing a full diagonalization of the nuclear spin Hamiltonian. The intensity of an ENDOR transition was assumed to be proportional to that of the EPR transition. A more detailed description of the simulation program is available in [36]. For a nucleus with I=1/2, six parameters are required to specify the hyperfine tensor: the isotropic hfc (aiso), two anisotropic hfc’s, and furthermore three Euler angles, relating the orientation of the principal axes of the hyperfine tensor to that of the g-tensor. Initial values for the Euler angles were calculated as follows. The coordinates of the β-CH2 protons of all coordinating cysteines in the X-ray structure of the reduced enzyme [18] were calculated. Then the Ni-H vectors were calculated, which were taken as a rough estimate for the direction of the dipolar hyperfine axis. These directions were transformed from the crystallographic axes system into the g-tensor axes system by making use of our knowledge of the complete g-tensor from single crystal EPR spectroscopy [23]. The simulations were subsequently optimized by fitting the six parameters to the signals observed in the experimental ENDOR spectra, whereby each ENDOR transition was assigned a fixed line width of 380 kHz in the simulation. Following the optimization, the hyperfine tensors were transformed back to the crystallographic axes system. It should be noted that the principal axes of the hyperfine tensors can be rotated by 180° about the g-tensor principal axes without affecting the simulation. This means that there are four possible choices for the directions of the axes, related by 180° rotations around the principal axes of the g-tensor, which give identical ENDOR simulations.
Simulations of the HYSCORE spectra were performed using a simulation program previously employed for ESEEM simulations [37] in which the HYSCORE modulation formula [33] was incorporated. This performs a full diagonalization of the nuclear spin Hamiltonian. When more than one nuclear spin interacts with the electron spin, the nuclear spin Hamiltonian is diagonalized separately for each nucleus and the modulation function is obtained from the product rule [38]. The amplitudes are calculated from the eigenvectors of the nuclear spin Hamiltonian. The program computes the spectrum at a given magnetic field and microwave frequency by considering only those molecules for which the orientation is such that the effective g value fulfills the resonance condition. This is the case when the effective g value of the particular orientation and the g value determined by the fixed magnetic field and microwave frequency are close to each other. In this procedure, the linewidth parameters of the EPR spectrum are used as a criterion, where it was assumed that the linewidth tensor is collinear with the g-tensor. Then, for each nucleus, six parameters are needed to specify the hyperfine tensor, and for nuclei with I>1/2, five additional parameters for the (traceless) quadrupole tensor. For deuterium (in the bridging position between Ni and Fe), initial values for the hyperfine parameters aiso and dipolar part of the anisotropic tensor (adip) were determined from the HYSCORE spectra according to the method described by Pöppl et al. [39]. This procedure is feasible since three out of four 2H cross peaks do not overlap with other signals. For nitrogen, initial values for the quadrupole parameters e2 qQ/h and η can be derived from the frequencies that correspond to the nuclear quadrupole transitions (see below). The transitions were assigned a linewidth of 110 kHz. After considering all possible orientations (an equally-spaced grid with 200 steps for the polar angle θ was used), the 2-D array can be directly compared with the experimental spectrum.
For 14N, strong modulations are observable when the nuclear Zeeman and hyperfine terms cancel each other in one MS manifold (|A|=2νn). Then, only the nuclear quadrupole transitions ν+, ν−, ν0 are observed (ν+ = ν−+ν0), yielding sharp lines with strong intensities. They are related to the principal values of the traceless quadrupole tensor, expressed by the parameters K and η: K = e2qQ/4h = (ν++ν−)/6, η = 3ν0/(ν++ν−). From the other MS manifold a (broader) double quantum transition νDQ is usually observed from which the effective hfc can be obtained: νDQ = 2 [(ν n ±| A|/2)2+ξ2]1/2 with ξ =K(3+η2)1/2 [38, 40]. In HYSCORE spectra, correlation frequencies are expected for 14N between νDQ and any of the three frequencies (ν0, ν+, ν−) of the other MS manifold.
Simulations are performed for the HYSCORE part (off-diagonal part) of the spectrum only. No attempts have been made to simulate the signals on the diagonal, which result from the stimulated (three pulse) echo and incomplete population inversion by the microwave π pulse.
Results
EPR spectroscopy
The continuous-wave (cw) EPR spectra for the reduced Ni-C state of D. vulgaris Miyazaki F hydrogenase in H2O and in D2O measured at 50 K are shown in Fig. 2a, b respectively. Simulations of the spectra are also included and simulation parameters are given in Table 1. The Ni-C state is characterized by g values that are compatible with those found by other groups for similar [NiFe]-hydrogenases [20, 21, 26, 30, 41, 42] and characteristic for a Ni(III) state where the unpaired electron is mainly in a \( d_{{z^{2} }} \) orbital of nickel. A narrowing of the linewidths at all canonical orientations is observed upon solvent exchange to D2O and subsequent reduction of the sample with D2. The most pronounced narrowing is present at the g y orientation, for which the linewidth (peak-to-peak) reduces from 2.15 to 1.67 mT. This line-narrowing largely results from the exchange of a hydride directly bound to the [NiFe] center, as was deduced from earlier observations with ENDOR and HYSCORE spectroscopy [20, 22], the magnitude of the hyperfine coupling being on the order of 20 MHz. A similar line-narrowing upon D2O exchange has recently been observed by Müller et al. [26] for D. gigas hydrogenase.

X-band EPR spectra of the Ni-C state of the [NiFe]-hydrogenase of D. vulgaris Miyazaki F at T=50 K in H2O (a) and D2O (b). The principal values of the g-tensor are indicated. The simulations (blue lines) were obtained by using the parameters in Table 1. Experimental conditions: νmw=9.60 GHz, Pmw=1.0 mW, modulation amplitude=0.28 mT
When the proximal [4Fe4S] cluster is paramagnetic and the temperature is lowered to below 40 K, the Ni-C EPR signal broadens. Below 12 K the signal splits, which is a result of spin–spin interaction between the [NiFe] and proximal [4Fe4S] centers [30, 43]. The splitting of the Ni-C EPR signal can be largely avoided by setting the redox potential such that the [4Fe4S] clusters are oxidized. The mid-point potential of the Ni-C state happens to be close to that of the [4Fe4S] clusters of the small subunit [6, 29, 44, 45]. For D. vulgaris Miyazaki F hydrogenase the redox potential was carefully chosen such that 50–70% of the [4Fe4S] clusters were in the oxidized state (see “Materials and methods” section).
Orientation-selected ENDOR spectroscopy
Initial cw and pulse ENDOR experiments on samples which exhibit a completely split Ni-C EPR spectrum were not successful. No ENDOR effect was observed because of the enhanced relaxation rate caused by the interaction with the paramagnetic proximal [4Fe4S] cluster. By making use of carefully prepared Ni-C samples in which the potential was set such that the [4Fe4S] clusters remained largely oxidized, unsplit Ni-C EPR signals could be obtained in high quantity (50–70%). With a ratio of split:unsplit Ni-C of roughly 1:2, cw-ENDOR signals could be observed. However, pulse ENDOR signals remained difficult to obtain, most probably because the nuclear relaxation is still faster than the timescale of the pulse ENDOR experiment. The cw-ENDOR effect of this sample is weak and only high enzyme concentrations (~2 mM) and prolonged accumulation times (~3–12 h) led to acceptable signal-to-noise ratios. The optimal signal-to-noise ratio was obtained at a temperature of 25 K.
A set of orientation-selected ENDOR spectra for the hydrogenase in H2O is shown in Fig. 3. The effective g value is indicated to the right of each spectrum. The spectra reveal an intense transition at the 1H Zeeman frequency, νH, which results from protons relatively distant from the [NiFe] center. Signals of lower intensity are observed at frequency shifts of up to 9 MHz. Almost all prominent signals are symmetrically displayed around νH, with the signals on the high-frequency side of νH being more intense than those observed on the low-frequency side. The range of frequency shifts observed here is comparable to that observed in an earlier study by Müller et al. [26] on D. gigas [NiFe]-hydrogenase. Because of a better signal-to-noise ratio in their spectra, Müller et al. were able to observe more signals in this region.

Top: field-frequency plot of the experimental Ni-C ENDOR spectra. The colored lines connect signals that belong to proton a (blue), b (red) or c (green). Bottom: orientation selected cw-ENDOR spectra for the Ni-C state of D. vulgaris Miyazaki F hydrogenase recorded at various effective g values. Simulations of the ENDOR spectra are indicated by blue lines, see text and Table 2. Experimental conditions: νmw=9.5 GHz, T=25 K, Pmw=30.2 mW, RF frequency modulation 12.5 kHz, modulation amplitude 200 kHz, accumulation time for the individual spectra varies between 3 and 12 h, depending on the magnetic field setting
For the analysis of the ENDOR spectra, a set of transitions had to be attributed to one distinct nucleus by tracing the positions of the transitions over the whole field range. In this procedure, signals from one proton have to be grouped together, which can be facilitated by means of a field-frequency plot (see Fig. 3, top). The three largest hyperfine couplings can be clearly distinguished in the ENDOR spectra of Ni-C. The signals belonging to these couplings are named a, b and c.
In a next step, the ENDOR signals were simulated. As starting parameters for the simulations, the maximum (A||) and minimum (A⊥) shift of the signals were read from the field-frequency plot. Using the axial hyperfine tensor to start with, the isotropic hyperfine coupling constant (aiso = 1/3(A|| + 2A⊥) and purely dipolar hfc A⊥ ′ = 1/3(A⊥ − A||) were calculated. For the initial simulations, the orientation of the dipolar axes is also needed. For this purpose the positions of all β-CH2 protons of the coordinating cysteine ligands were deduced using the X-ray structure of the reduced enzyme [18] and the Ni-H vectors were calculated as first estimates for the directions of the dipolar axes. Transformation of the vectors into the g-tensor principal axis system [23] yielded a set of Euler angles relating the directions of the dipolar axes to the principal g-tensor axes. Then it was tested which of the dipolar axes orientations, with the appropriate initial hyperfine parameters, reproduced the signals in the field-frequency plot best. After subsequent variation of all the parameters—first optimizing the simulation at the single crystal-like positions, g x and g z , and then for the intermediate field positions, and finally relaxing the restriction of axially symmetric hyperfine tensors—a good simulation of the ENDOR spectra and an estimate for the errors of the individual adjustable parameters was obtained. The resulting simulations are included in Fig. 3 (bottom) and the hyperfine coupling constants are summarized in Table 2. The ENDOR simulations are insensitive to 180° rotations of the principal axes of the hyperfine tensor about any of the principal axes of the g-tensor.
It is apparent from Fig. 3 that the signals assigned to protons a, b and c are well simulated. Furthermore, the signals of protons a and b have the largest frequency shift and they behave similarly when the magnetic field setting is changed. From the simulation of these signals relatively large isotropic hyperfine interactions of 13.7 and 11.8 MHz are elucidated, which is an indication that these signals do indeed originate from the β-CH2 protons of one cysteine residue carrying a large fraction of the spin density. The signals of proton c have a smaller shift than those of protons a and b. Proton c is also characterized by a hyperfine tensor, which is dominated by the isotropic coupling constant (8.9 MHz), and the anisotropic part is comparable in size to those of protons a and b.
Additionally, experiments were performed on samples in D2O/D2 and orientation-selected ENDOR spectra were recorded at 17 magnetic field positions. The spectra (data not shown) looked very similar to those in H2O/H2 with only minor intensity changes. The signals of protons a, b and c are clearly observable in these spectra, indicating that these protons are not exchangeable.
HYSCORE spectroscopy
Figure 4a, b depict the HYSCORE spectra of the Ni-C state of D. vulgaris Miyazaki F hydrogenase in H2O and D2O in the low-frequency regime. The spectra are dominated by strong signals on the diagonal, which stem from the stimulated (three pulse) echo. In the spectrum in H2O, three signals at 0.5, 1.2 and 1.7 MHz are present, indicated with blue dots. Such a pattern, where the two smallest frequencies add up to the largest frequency, indicates that these signals stem from one nucleus with I=1 (nitrogen). Under the “exact cancellation” condition [40], the nuclear Zeeman interaction and hyperfine interaction cancel each other in one of the MS manifolds and the three frequencies ν+, ν− and ν0 are determined by the quadrupole interaction of this nucleus. Additional signals on the diagonal are found at 2.3 and 3.5 MHz. At 3.9 MHz an artifact from the spectrometer related to the 125 MHz clock frequency of the pulse programmer appears. Cross peaks of lower intensity are observed in the spectrum in H2O, which relate the nitrogen single-quantum frequencies of 1.2 and 1.7 MHz to a broad double-quantum transition between 4 and 5 MHz (cross peak labeled (i), refer to Fig. 1). Cross peaks (ii) also relate the peak at 2.3 MHz to about 3.9 MHz. These frequencies probably also stem from nitrogen (vide infra).

HYSCORE spectra (14N/2H region) for the Ni-C state of D. vulgaris Miyazaki F hydrogenase in H2O (a) and D2O (b), recorded at the g y canonical orientation (B0=325.0 mT). Experimental conditions: νmw=9.7 GHz, T=3.1 K, length of 90° pulse 16 ns, τ1=120 ns, τ2=160 ns, τ3=200 ns (addition of three spectra). Steps of t1 and t2 16 ns, resolution 256×256 data points, accumulation time ~5 h per spectrum. Also included is a HYSCORE spectrum of the reduced [4Fe4S] clusters in H2O (c), recorded at B=345.1 mT, τ=120 ns. All signals in a belong to nitrogen, while in b additional signals from 2H (indicated in red) are superimposed. The blue dots in a indicate intense lines on the diagonal belonging to zero field frequencies ν0, ν−, ν+ of the 14N. In a the labels i and ii indicate cross peaks between νDQ and one of the single quadrupole frequencies ν+ and ν− (see text)
Upon H2O → D2O exchange of the solvent, additional cross peaks become visible in Fig. 4b. These cross peaks are only marginally above the noise level (indicated in red in the figure). The first one lies close to the Zeeman frequency of deuterium (2.1 MHz) and extends up to 1 MHz away from the diagonal. A similar peak, albeit one with even less intensity, is present at exactly double the frequency. In addition, a “v” shaped cross peak appears, which starts at approximately 3.2 MHz and ends at (5.5, 2.6) MHz and (2.6, 5.5) MHz. Note that the relaxation properties of the spin system reacted sensitively on H2O→D2O solvent exchange, and the nitrogen couplings are more pronounced in the HYSCORE spectra of the D2O sample. Differences in relaxation rates have been observed before in ENDOR spectra of hydrogenases [20, 21].
For comparison, an additional HYSCORE spectrum was recorded at a magnetic field setting where only the [FeS] clusters contribute to the EPR spectrum (Fig. 4c). It should be clearly noted that the [4Fe4S] clusters, when they are reduced as is the case for split Ni-C, give rise to a pulse EPR signal which overlaps with that of the [NiFe] center over the entire field-range spanned by the g values of Ni-C. The HYSCORE spectrum for the [FeS] clusters was recorded at a magnetic field setting which is marginally larger than the one that corresponds to the g z value of the Ni-C state. In this spectrum, the strong diagonal peaks indicated by the blue dots disappeared, indicating that they belong to a nitrogen associated with the [NiFe] center, but the intensity remains in the area between 2.5 and 4.5 MHz. A pattern like this, where only the nuclear double-quantum transitions of both MS manifolds are present, is indicative of a nitrogen for which the exact cancellation condition [40] is not fulfilled. As both the proximal and distal [4Fe4S] cluster are coordinated by a histidine (His 235 for the proximal cluster and His 188 for the distal cluster), it is likely that these signals are related to these residues and not to a nitrogen near the [NiFe] center. Lastly, the signals indicated in red in the spectrum in D2O (Fig. 4b) have also disappeared in Fig. 4c, indicating that these signals are related to the [NiFe] center. The pattern marked with the blue dots at 0.5, 1.2 and 1.7 MHz (Fig. 4a) is indicative of a histidine nitrogen [46, 47] and we therefore assign this pattern to the ε nitrogen of histidine-88 of the large subunit, which forms a hydrogen bond to the Cys-549 of the [NiFe] center.
In a next step, the signals associated with the [NiFe] center (the nitrogen and deuterium signals) were simulated. A single HYSCORE spectrum recorded at g y and τ=200 ns is shown in Fig. 5a. The deuterium signals are very similar to those found recently for the Ni-C state of the regulatory hydrogenase of R. eutropha [22]. The latter enzyme has the advantage that the [4Fe4S] clusters remain oxidized when the [NiFe] center is in the Ni-C state, and experiments at low temperatures are therefore possible due to the absence of the spin–spin interaction of the [NiFe] center and the proximal [4Fe4S] cluster. Furthermore, the histidine, which forms a hydrogen bond to one of the Ni-coordinating sulfurs in D. vulgaris, is not present in this enzyme, and therefore nitrogen contributions are absent in the HYSCORE spectrum of R. eutropha. The HYSCORE spectrum of this enzyme in D2O, recorded under the same conditions, is reproduced in Fig. 5b. It is apparent that the signal-to-noise ratio is much better than that of Fig. 5a. Nevertheless, the deuterium signals can clearly be seen in both spectra. Cross peak I corresponds to correlations between the single-quantum transitions of both MS manifolds, cross peak II (the “v” shaped cross peak in Fig. 4) to correlations between a single quantum transition of one MS manifold with the double quantum transition of the other MS manifold, and cross peak III to correlations between the double quantum transitions of both MS manifolds.

HYSCORE spectra (14N/2H region) for a Ni-C of D. vulgaris Miyazaki F hydrogenase in D2O at the g y canonical orientation (νmw=9.7 GHz, τ=200 ns, B0=325.0 mT, length of the 90° pulse 16 ns), and b Ni-C of the regulatory hydrogenase of R. eutropha, reproduced from [22]. Also included is a simulation for a nitrogen nucleus, assigned to the Nε of histidine-88 [47] (c) and a simulation for the deuterium bridging ligand (d) (see [22]). The simulation for the deuterium bridging ligand in D. vulgaris Miyazaki F, which is very similar to that of R. eutropha is superimposed in gray in a
Although the signal-to-noise ratios differ, the deuterium signals in Fig. 5a could be analyzed and are found to be almost the same as in Fig. 5b. Therefore, HYSCORE simulations were performed with the same parameters as those used for the deuterium nucleus of R. eutropha. The parameters are given in Table 3 and the simulation is shown in Fig. 5d, where it is seen that all deuterium-related signals of the experimental spectra are reproduced well by the simulations. It should be noted that the quadrupole splitting for deuterium (≲100 kHz) [22] is smaller than the observed linewidth. In order to keep the number of parameters to a minimum, the quadrupole interaction has therefore been neglected in the simulation.
Since the correlation frequencies of the nitrogen are poorly resolved in the experimental spectra, severe limitations have been placed on the number of free parameters in the simulation procedure. First, from the three single-quantum frequencies on the diagonal (see Fig. 4a), the quadrupole parameters have been estimated using the formulae given in “Materials and methods”. The double quantum frequency of nitrogen is broad and spans a range from 4 to 5 MHz, as seen from the signal on the diagonal at 4.5 MHz and from the cross peaks at (4.5, 0.5) MHz and (1.6, 4.5) MHz. From this, starting values for the isotropic hyperfine coupling constant aiso and dipolar constant A x ′ were obtained. The hyperfine tensor was restricted to being axial. The principal axes were kept fixed to the following directions based on the crystal structure [18]: the x-axis of the hyperfine tensor (dipolar axis) was chosen parallel to the Nε-H (His 88) direction, which also points to the large spin density on the Sγ of Cys-549 [25]. The quadrupole x-axis was also parallel to the Nε-Hε direction, the y-axis perpendicular to the imidazole plane, and the z-axis was tangential to the imidazole [46]. With these initial parameters, the simulations were optimized by changing only aiso, A x ′ and e2 qQ/h. Optimized parameters are given in Table 4 and the resulting simulation is shown in Fig. 5c. The modulation depth is similar to that of the deuterium nucleus (Fig. 5d), but for a better comparison with the experimental spectrum, the contour levels have been chosen differently.
The simulation shows 12 cross peaks (Fig. 5c). Unlike for the deuterium, these cross peaks are directed either horizontally or vertically in the spectrum. The reason for this is that the single quantum transitions of the cancelled MS manifold are largely determined by the nuclear quadrupole interaction, which does not vary with field. In general, the presence of horizontally - or vertically - oriented cross peaks in the low-frequency region of HYSCORE spectra is indicative of nitrogen. The six horizontal (or vertical) cross peaks correlate the single quantum frequencies in the cancelled MS manifold with the double quantum frequency of the non-cancelled manifold [at (0.5, 4.4) MHz, (1.2, 4.4) MHz and (1.7, 4.4) MHz], and the single quantum frequencies in the cancelled manifold to the single quantum frequencies of the non-cancelled MS manifolds [at (0.5, ~2.0) MHz, (1.2, ~2.0) MHz and (1.7, ~2.0) MHz]. Although good agreement cannot be expected due to the low signal-to-noise ratio of the experimental spectrum, the most intense cross peaks of the simulation (Fig. 5c) can easily be found in Fig. 5a.
Discussion
In the discussion, we first focus on the assignment of the signals in the ENDOR spectra to non-exchangeable protons of cysteine ligands of the [NiFe] center. Secondly, the signals in the HYSCORE spectrum in D2O will be assigned based on comparison with data for the Ni-C state of the regulatory hydrogenase of R. eutropha [22].
Assignment of the non-exchangeable protons
In the field-frequency plot (Fig. 3, top) contributions from three protons (labeled a, b and c) are observed. Additional experiments following D2O exchange indicate that these protons belong to non-exchangeable protons. The shape of the curves which connect the resonances from the same proton are virtually identical to those observed earlier for D. gigas and D. baculatum hydrogenase [26]. In agreement with conclusions drawn in this work, our data confirm that the [NiFe] center is essentially the same in these enzymes, even up to the detailed nuclear resolution that can be addressed by orientation-selected ENDOR spectroscopy. The protons a, b and c of this work correspond well to protons named H-A, H-B and H-C in [26]. The large isotropic hyperfine interactions show that these protons belong to β protons of the Ni-coordinated cysteines. Nevertheless, upon critical comparison, subtle differences can be found. For example, the field-frequency plot of D. gigas hydrogenase [26] reveals that the largest hyperfine coupling constant for the most strongly coupled proton (H-A) is about 18.5 MHz, whereas the plot for D. vulgaris hydrogenase (Fig. 3, top) shows that this constant is 17.9 MHz. Differences of the order of ≲ 1 MHz are also found for the other hyperfine coupling constants determined from the simulations.
In order to assign the protons a, b and c to any of the β protons of the Ni-coordinating cysteines, the directions of the principal axes of the hyperfine tensor with respect to those of the g-tensor are required. The directions follow directly from the simulation of the ENDOR spectra. We will mainly focus on the direction of the “dipolar axis”, which corresponds to the largest principal value (the z-axis of the tensors of protons a, b and c). This direction is approximately parallel to the direction, which connects the location of the bulk spin-density (on nickel) and the position of the proton. Therefore, by using Ni-H starting vectors in the simulation program and combining them with knowledge of the g-tensor of the Ni-C state of D. vulgaris hydrogenase [23], good agreement was quickly found. The angle between the dipolar axis of proton a and the Ni-H vector for one of the β-protons of Cys-549 is 7°, the angle for proton b and the Ni-H vector of the other β-proton of Cys-549 is 6°, and the one for proton c and the Ni-H vector of a proton of Cys-546 is 12°. We therefore assign protons a, b and c to these β protons, which is also intuitively expected since protons a and b show a similar behavior in the field-frequency plot (Fig. 3, top).
In Table 5, a detailed comparison is given between the hyperfine coupling parameters for D. vulgaris Miyazaki F and D. gigas hydrogenase, including the directions of the principal axes, which directly follow from the simulations. The simulations are, however, insensitive to 180° rotations of the hyperfine axes around any of the g-tensor axes (see Scheme 1 and “Materials and methods”). Neglecting the absolute signs of the direction cosines for a moment for the above reason, the largest angle between the dipolar axes for D. vulgaris and D. gigas hydrogenase occurs for proton a and amounts to 30°, which is about equal to the combined accuracy of both experiments. The angles are included in Table 5. We therefore conclude that the directions of the dipolar hyperfine axes of D. vulgaris and D. gigas hydrogenase, neglecting the absolute signs of the direction cosines, are similar. When our signs are taken for the direction cosines (see “Appendix”) of the hyperfine axes of D. gigas hydrogenase [26], a similar assignment is obtained for this enzyme as well. In this case, the results from single crystal EPR data [23], from the ENDOR data presented here and in reference [26], and the DFT calculations for the Ni-C state are all in agreement. This result shows that the overall electronic structure of the [NiFe] center in the active Ni-C state is essentially the same in all [NiFe]-hydrogenases.

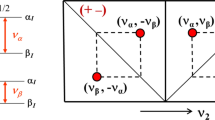
Illustration of the sign-ambiguity problem in ENDOR spectroscopy. The directions of the principal axes of the g tensor (with the z-axis chosen perpendicular to the plane of the paper) are shown in black, and those of the hyperfine tensor in magenta (for simplicity the hyperfine z-axis is also perpendicular to the plane of the paper). Other orientations of the hyperfine tensor, which give exactly the same field-frequency dependence in ENDOR spectroscopy, can be obtained by 180° rotations about any of the g tensor axes and are indicated in red, yellow and blue. It is then clear that the angles with the metal-ligand direction (indicated as M–L; for example Ni–H) can be different for all four possibilities
Assignment of the exchangeable proton
The deuterium signals observed in the HYSCORE spectrum of D. vulgaris Miyazaki F (see Fig. 5a) look the same as those for the regulatory hydrogenase of R. eutropha (Fig. 5b). This is the first observation of these signals for a catalytically active hydrogenase. The regulatory hydrogenase does not exhibit split Ni-C signals and a hydrogen-bonded histidine to one of the Ni coordinating cysteines is absent. The simulation presented in Fig. 5d is performed with the hyperfine parameters of R. eutropha hydrogenase. Since the analysis is identical to the one presented for the regulatory hydrogenase, we refer to [22] for details. These signals were assigned to a formal hydride (1H−/2H−) located in the bridging position between Ni and Fe [22, 23]. The signals in Fig. 5a show that a hydride bridge is also present in the Ni-C state of the catalytic hydrogenase of D. vulgaris Miyazaki F. The hydride is bound in the equatorial plane of the \( d_{{z^{2} }} \) orbital, which explains the small and negative isotropic hyperfine coupling constant; a result that is in full agreement with DFT calculations [25], see Table 6. Further smaller deuterium couplings are not resolved due to the presence of nitrogen signals in the same frequency region. The observation of the deuterium HYSCORE signals in both a regulatory and a catalytic hydrogenase suggests that the electronic structures of the [NiFe] centers in these proteins are also very similar and that differences in their activity most probably arise from effects located further away from the [NiFe] center.
Summary and conclusion
Detailed information about the spin density distribution of the [NiFe] center has been obtained by applying both orientation-selected ENDOR and complementary HYSCORE spectroscopy to [NiFe]-hydrogenase frozen solutions. Knowledge of the orientation of the g-tensor, determined earlier through EPR studies of hydrogenase single crystals [23], was a prerequisite for the analysis.
The detection of H/D-exchangeable deuterium signals in the HYSCORE spectra of the Ni-C state of hydrogenase, as found earlier in a regulatory hydrogenase [22], provides direct experimental evidence for the presence and the binding mode (bridging) of a hydride at the Ni site of catalytic [NiFe]-hydrogenases, which was predicted earlier from comparison of single crystal EPR data and DFT calculations [23].
The complete hyperfine coupling tensors for three distinct protons of the Ni-C state of the hydrogenase from D. vulgaris Miyazaki F have been determined from orientation-selected ENDOR spectroscopy. These three couplings are attributed to β-CH2 protons of the Cys-549 and one β-CH2 proton of Cys-546, which are consistent with earlier predictions by DFT [25]. Slight deviations between different hydrogenases could be detected and were assigned to small structural differences.
Furthermore, hydrogenase nitrogen couplings have been detected in the HYSCORE spectra of the D. vulgaris. These were assigned to the Nε of His-88, which forms a hydrogen bond to Sγ(Cys-549). The data analysis and assignment is in full agreement with the detailed study of Buhrke et al. [47] on Ni-C in R. eutropha and Chapman et al. [48] on Ni-C in D. gigas. Additional nitrogen couplings associated with the [4Fe4S] clusters have been observed. Although the nuclear quadrupole constants could not be determined, these nitrogens are also assigned to His ligands present in the vicinity of the FeS clusters.
Comparison with experimental and theoretical data available for other [NiFe]-hydrogenases reveals a consistent picture in which the z axis of the \( 3d_{{z^{2} }} \) orbital on Ni is directed towards Sγ(Cys-549) and the free coordination position of Ni. Moreover, the data show that the [NiFe] centers in catalytically-active and regulatory hydrogenases are essentially the same. This suggests that the functional differences of these hydrogenases originate from structural differences further away from the [NiFe] center.
References
Frey M (1998) Struct Bond 90:97–126
Vignais PM, Billoud B, Meyer J (2001) FEMS Microbiol Rev 25:455–501
Albracht SPJ (1994) Biochim Biophys Acta 1188:167–204
Moura JJG, Moura I, Huynh B-H, Krüger H-J, Teixeira M, DuVarney RC, DerVartanian DV, Xavier AV, Peck HD Jr, LeGall J (1982) Biochem Biophys Res Commun 108:1388–1393
Fernandez VM, Hatchikian EC, Patil DS, Cammack R (1986) Biochim Biophys Acta 883:145–154
Coremans JMCC, van Garderen CJ, Albracht SPJ (1992) Biochim Biophys Acta 1119:148–156
Krasna AI (1978) Method Enzymol 53:296–304
van der Zwaan JW, Albracht SPJ, Fontijn RD, Slater EC (1985) FEBS Lett 2:271–277
van der Zwaan JW, Albracht SPJ, Fontijn RD, Roelofs YBM (1986) Biochim Biophys Acta 872:208–215
Higuchi Y, Yagi T, Yasuoka N (1997) Structure 15:1671–1680
Volbeda A, Charon M-H, Hatchikian EC, Frey M, Fontecilla-Camps JC (1995) Nature 373:580–587
Volbeda A, Garcin E, Piras C, De Lacey AL, Fernandez VM, Hatchikian EC, Frey M, Fontecilla-Camps JC (1996) J Am Chem Soc 118:12989–12996
Rousset M, Montet Y, Guigliarelli B, Forget N, Asso M, Bertrand P, Fontecilla-Camps JC, Hatchikian EC (1998) Proc Natl Acad Sci USA 95:11625–11630
Matias PM, Soares CM, Saraiva LM, Coelho R, Morais J, LeGall J, Carrando MA (2001) J Biol Inorg Chem 6:63–81
Stein M, van Lenthe E, Baerends EJ, Lubitz W (2001) J Am Chem Soc 123:5839–5840
Stadler C, De Lacey AL, Montet Y, Volbeda A, Fontecilla-Camps JC, Conesa JC, Fernandez VM (2002) Inorg Chem 41:4424–4434
Carepo M, Tierney DL, Brondino CD, Yang TC, Pamplona A, Telser J, Moura I, Moura JJG, Hoffman BM (2002) J Am Chem Soc 124:281–286
Higuchi Y, Ogata H, Miki K, Yasuoka N, Yagi T (1999) Structure 7:549–556
Garcin E, Vernede X, Hatchikian EC, Volbeda A, Frey M, Fontecilla-Camps JC (1999) Structure 7:557–566
Fan C, Teixeira M, Moura JJG, Moura I, Huynh B-H, LeGall J, Peck HD Jr, Hoffman BM (1991) J Am Chem Soc 113:20–24
Whitehead JP, Gurbiel RJ, Bagyinka C, Hoffman BM, Maroney MJ (1993) J Am Chem Soc 115:5629–5635
Brecht M, van Gastel M, Buhrke T, Friedrich B, Lubitz W (2003) J Am Chem Soc 125:13075–13083
Foerster S, Stein M, Brecht M, Ogata H, Higuchi Y, Lubitz W (2003) J Am Chem Soc 125:83–93
Stein M, Lubitz W (2001) Phys Chem Chem Phys 3:2668–2675
Stein M, Lubitz W (2001) Phys Chem Chem Phys 3:5115–5120
Müller A, Tscherny I, Kappl R, Hatchikian EC, Hüttermann J, Cammack R (2002) J Biol Inorg Chem 7:177–194
Yagi T, Kimura K, Daidoji H, Sakai F, Tamura S, Inokuchi H (1976) J Biochem 79:661–671
Lide DR (ed)(1992) CRC Handbook of chemistry and physics. Boca Raton, FL
Cammack R, Patil DS, Hatchikian EC, Fernandez VM (1987) Biochim Biophys Acta 912:98–109
Guigliarelli B, More C, Fournel A, Asso M, Hatchikian EC, Williams R, Cammack R, Bertrand P (1995) Biochemistry 34:4781–4790
Zweygart W, Thanner R, Lubitz W (1994) J Magn Reson Ser A 109:172–176
Höfer P, Grupp A, Nebenführ H, Mehring M (1986) Chem Phys Lett 132:279–282
Shane JJ, Höfer P, Reijerse EJ, de Boer E (1992) J Magn Reson 88:241–256
Gemperle C, Aebli G, Schweiger A, Ernst RR (1990) J Magn Reson 88:241–256
Fahnenschmidt M (2000) PhD Thesis, TU Berlin, Germany http://edocs.tu-berlin.de
Geßner C (1996) PhD Thesis, TU Berlin, Germany
van Gastel M, Coremans JWA, Jeuken LJC, Canters GW, Groenen EJJ (1998) J Phys Chem A 102:4462–4470
SA Dikanov, Tsvetkov YD (1992) Electron spin echo envelope modulation (ESEEM) spectroscopy. CRC, Boca Raton, FL
Pöppl A, Kevan L (1996) J Phys Chem 100:3387–3394
Flanagan HL, Singel DJ (1987) J Chem Phys 87:5606–5616
Sorgenfrei O, Klein A, Albracht SPJ (1993) FEBS Lett 332:291–297
van der Zwaan JW, Coremans JMCC, Bouwens ECM, Albracht SPJ (1990) Biochim Biophys Acta 1041:101–110
Dole F, Medina M, More C, Cammack R, Bertrand P, Guigliarelli B (1996) Biochemistry 35:16399–16406
De Lacey AL, Hatchikian EC, Volbeda A, Frey M, Fontecilla-Camps JC, Fernandez VM (1997) J Am Chem Soc 119:7181–7189
Coremans JMCC, van der Zwaan JW, Albracht SPJ (1989) Biochim Biophys Acta 997:256–267
Jiang F, McCracken J, Peisach J (1990) J Am Chem Soc 112:9035–9044
Buhrke T, Brecht M, Lubitz W, Friedrich B (2002) J Biol Inorg Chem 7:897–908
Chapman A, Cammack R, Hatchikian EC, McCracken J, Peisach J (1988) FEBS Lett 242:134–138
Acknowledgements
The authors are grateful to Matthias Stein for performing the DFT calculations. Yoshiki Higuchi (Hyogo University, Japan) kindly provided the hydrogenase samples of D. vulgaris Miyazaki F. This work has been supported by Deutsche Forschungsgemeinschaft (Sfb 498, TP C2), Fonds der chemischen Industrie (WL), and by the Max Planck Society.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Choosing signs for the direction cosines from the ENDOR spectra for D. gigas hydrogenase
When analyzing orientation-selected ENDOR spectra, information is extracted in the form of principal values of the hyperfine tensor and direction cosines. However, the absolute sign of the direction cosines cannot be determined from analysis of the experimental data.
Müller et al. [26] used their choice of signs for the direction cosines to reconstruct the orientation of the g-tensor for Ni-C in D. gigas hydrogenase. We propose a different sign choice for the following reasons. (1) By inspecting the field-frequency plot (see Fig. 3, top), protons a and b are found to be the two most strongly coupled protons and display a similar field-dependence, whereas the curve for proton c looks different. This is a strong indication that protons a and b (or H-A and H-B for D. gigas hydrogenase) belong to the same cysteine residue (and likewise protons H-C and H-D in [26]). (2) The assignment of Müller disagrees with the spin-density distribution calculated by two independent groups [16, 24, 25], which indicates that the sulfur of Cys-549 (Cys-533 for D. gigas) carries significant spin-density, so the protons of this residue should have the largest hyperfine coupling constants. Calculated proton hyperfine coupling constants from [25] are given in Table 6 for comparison. The calculated spin density distributions show that the principal z-axis of the g-tensor is directed from nickel towards the vacant coordination position, which is also expected based on ligand field theory. Lastly, the direction cosines of the x axis of proton H-B suggest that the minimum hyperfine coupling of 10.329 MHz (see Table 5) is attained for a molecule oriented such that the effective g value is [(2.194×0.283)2 + 2.146×0.631)2 + (2.011×0.723)2]1/2=2.08. Inspection of the field-frequency plot in [26], however, reveals that proton H-B attains its minimum hyperfine coupling at g ≈2.15. The direction cosines of proton H-B seem to be in disagreement with the corresponding curve in the field-frequency plot of [26]. When the data for D. gigas hydrogenase [26] are used with the sign convention and assignment as chosen in the present work, a similar picture is obtained for the Ni-C state in D. gigas hydrogenase. Probably, the g-tensor for the Ni-C state of D. gigas hydrogenase is similar to the one determined by single crystal EPR for D. vulgaris hydrogenase [23], which is also compatible with theoretical studies [16, 24, 25].
Rights and permissions
About this article
Cite this article
Foerster, S., Gastel, M.v., Brecht, M. et al. An orientation-selected ENDOR and HYSCORE study of the Ni-C active state of Desulfovibrio vulgaris Miyazaki F hydrogenase. J Biol Inorg Chem 10, 51–62 (2005). https://doi.org/10.1007/s00775-004-0613-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-004-0613-5