
Abstract
Iron removal from serum transferrin by various chelators has been studied by gel electrophoresis, which allows direct quantitation of all four forms of transferrin (diferric, C-monoferric, N-monoferric, and apotransferrin). Large cooperativity between the two lobes of serum transferrin is found for iron removal by several different chelators near physiological conditions (pH 7.4, 37 °C, 150 mM NaCl, 20 mM NaHCO3). This cooperativity is manifested in a dramatic decrease in the rate of iron removal from the N-monoferric transferrin as compared with iron removal from the other forms of ferric transferrin. Cooperativity is diminished as the pH is decreased; it is also very sensitive to changes in chloride ion concentration, with a maximum cooperativity at 150 mM NaCl. A mechanism is proposed that requires closure of the C-lobe before iron removal from the N-lobe can be effected; the “open” conformation of the C-lobe blocks a kinetically significant anion-binding site of the N-lobe, preventing its opening. Physiological implications of this cooperativity are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The transferrins are a family of iron-binding proteins that include serum transferrin, lactoferrin, ovotransferrin, and melanoferrin [1, 2]. Transferrins consist of a single polypeptide chain (MW ~80 kDa) that is composed of two distinct lobes of similar N-terminal and C-terminal fragments, designated as the N- and C-lobes. The two lobes are further divided into two domains, which form a cleft for binding of the iron atom. Each lobe contains one iron-binding site. The coordinating ligands in both lobes of transferrin are identical and consist of two tyrosine residues, an aspartate residue, and a histidine residue; the coordination sphere is completed by a bidentate carbonate anion. A distinguishing feature of all transferrins is the requirement of a synergistic anion for strong iron binding; bicarbonate functions as the physiological synergistic anion. The iron atom is six coordinate in a pseudooctahedral geometry with nearly identical bond lengths in both lobes and in different forms of transferrin. Both domains participate in the binding of iron and are drawn toward each other upon iron binding, promoting closure of the binding cleft. The “closed” and “open” protein conformations have been demonstrated crystallographically for ovotransferrin (Fig. 1) [3, 4]. This large conformational change is the gate that controls iron removal since the “closed” form prevents chelators from interacting with the iron-binding site.

The crystal structure (top) and cartoon (bottom) of diferric ovotransferrin (left) and apo ovotransferrin (right). The coordinates were obtained from the Protein Data Bank under the identifiers 1DOT and 1AOV, respectively
There is a high degree of sequence homology between the two lobes and between different species and different transferrins [5, 6, 7]. The bilobal nature of the transferrins is believed to originate from genetic duplication of a monolobal progenitor [8]. Bilobal transferrins were once thought to have originated with the evolutionary emergence of the phylum Chordata, which was necessitated by the evolution of a filtration kidney that would excrete a monolobal transferrin [9]. However, iron-binding proteins have been found in several arthropod species [6, 10, 11, 12, 13]. The genetic sequence for the cockroach and hawkmoth proteins has been determined; both share more than 30% of the same genes as vertebrate transferrin [6, 7]. Aisen and co-workers [7] have demonstrated that the iron-transport protein in cockroaches shares all the key features of the transferrins (in particular, the synergistic anion requirement), which authenticates that these proteins are members of the transferrin family. Thus, the evolution of bilobal transferrin must be pushed back to at least 400 million years ago, and the evolutionary advantage conferred by a bilobal transferrin must lie elsewhere, since arthropods lack a filtration kidney.
Is cooperativity of the lobes the evolutionary advantage? It has been suggested that cooperation between the lobes provides greater levels of control [13]. That cooperativity is the subject of this paper.
Materials and methods
Iron chelators
Tiron was purchased from Sigma and used as received after analysis confirmed the purity of the material. Pyrosphosphate was purchased from Strem. Desferrioxamine B was a gift of Salutar. Enterobactin and amonabactin were isolated and purified according to literature procedures [14, 15]. The other chelators used in this work are hexa-, tetra-, or bidentate ligands containing catecholate or hydroxypyridonate moieties [15].
Preparation of diferric transferrin
Human apotransferrin (81 mg, 1 μmol) (Sigma) was dissolved in 3 mL of Tris buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 20 mM NaHCO3). An iron nitrilotriacetate, Fe(NTA), solution was prepared by dissolving 45 μmol of FeCl3⋅6H2O and 90 μmol of nitriloacetic acid in 2 mL of 6 M HCl, adjusting the pH to 4.0 with NaOH solution, and diluting to 10 mL. To the apotransferrin was added 2.0 mL of the Fe(NTA) solution and the resulting solution was incubated at 37 °C overnight. Unbound iron was removed by passing the transferrin solution through a Sephadex G25-column (PD-10 column, Pharmacia) equilibrated and eluted with the Tris buffer. The solution was stored at 4 °C until use. Protein concentration was determined by measuring the absorbance at 280 nm using an extinction coefficient of 113,000 M−1 cm−1. The percentage of iron loading was estimated by measuring the absorbance at 470 nm using an extinction coefficient of 5000 M−1 cm−1.
Urea-polyacrylamide gel electrophoresis
The gel was prepared following a modified procedure of Makey and Seal [16]: 8.7 mL of acrylamide (3.3% C/30% T) was added to 14.4 g of urea and 8 mL of TBE buffer concentrated five times (0.5 M Tris, 50 mM boric acid, 8 mM EDTA, pH 8.4), and the volume was adjusted to 40 mL with water. The gel was polymerized with the addition of 400 μL 10% ammonium persulfate and 20 μL TMEDA (N,N,N′,N′-tetramethylenediamine). The electrophoresis apparatus used was a Protean II xi Cell (BioRad). Electrophoresis was carried out for 16 h at 100 V, and proteins were stained with Coomassie brilliant blue R-250 (Bio-Rad). Generally, each experiment was repeated three times.
Kinetics of iron removal from transferrin
Iron removal studies from diferric transferrin were carried out at 37 °C and pH 7.4 in Tris buffer (50 mM Tris, 150 mM NaCl, and 20 mM NaHCO3). A typical kinetic run was performed as follows: a stock solution of the appropriate chelator was prepared by dissolving the desired amount of chelator in 1 mL of Tris buffer, heating this solution and 700 μL of a 136 μM diferric transferrin solution to 37 °C, and allowing 30 min for temperature equilibration. At this point, 300 μL of the chelator solution was added to the transferrin solution (final volume=1 mL; final transferrin concentration=95 μM). Aliquots of 10 μL were taken every 10 min. The aliquots were immediately quenched by adding 10 μL of loading buffer (0.1 M Tris-borate buffer, pH 8.4, 2 mM EDTA, 10% glycerol, 0.2% bromophenol blue) and frozen. At the conclusion of the kinetic study, samples were thawed and loaded onto a 6 M urea-polyacrylamide gel.
Results
Transferrin delivers iron to cells through active transport by a receptor-mediated process [1, 2, 17, 18, 19]. Each serum transferrin molecule undergoes 100–200 cycles of iron binding, transport, and release during its lifetime [17]. However, this process is quite different than the release of iron from transferrin in serum to low molecular weight chelation. The ability to remove iron from transferrin at a practical rate is an important feature of potential chelation drugs. Because the two lobes of transferrin are not kinetically equivalent, there are four distinct forms of transferrin, depending on the amount and location of iron bound: iron bound to both lobes (diferric), iron bound to the C-lobe but not the N-lobe (C-monoferric), iron bound to the N-lobe but not the C-lobe (N-monoferric), and no iron bound (apo). The concentrations of all four forms of transferrin must be followed to directly measure the kinetics of iron removal from different forms of the protein. The general scheme for this process is shown in Scheme 1, where, for example, k1N refers to the rate at which iron is removed from the N-lobe in diferric transferrin and k2C refers to the rate at which iron is removed from the C-lobe in the C-monoferric form [20]. If there is cooperativity between the two lobes, then k1N≠k2N and/or k1C≠k2C. The magnitude of this difference will reflect the extent of cooperativity.

The removal of iron from transferrin by strong chelators has been commonly monitored by UV/Vis spectroscopy, which gives only the total amount of iron bound to Tf. In a few cases, gel electrophoresis has been utilized [14, 21, 22]. The advantage of gel electrophoresis is that the concentrations of all four forms of transferrin are determined as a function of time, directly determining the microscopic rate constants.
There are several factors that appear to affect iron removal, including (but not limited to) the chelator used, the concentration of chelating ligand, the ionic strength, the pH, the temperature, and the concentration and type of added salts [14, 15, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80]. Despite much study, a general mechanism for iron removal from human transferrin that accounts for all the observed kinetic behavior has remained elusive. Many studies employed one or more conditions quite different than those that would be encountered under physiological conditions (i.e. studying reactions at 25 °C or with no added NaCl or NaHCO3). The present study was performed near physiological conditions (37 °C, pH 7.4, 150 mM NaCl, 20 mM NaHCO3) and used several different ligands (ligand abbreviations and structures are shown in Fig. 2).

Structures and abbreviations of the chelating ligands used for iron removal from transferrin
A typical gel for iron removal from diferric transferrin by 3,2-HOPO is shown in Fig. 3. A buildup of N-monoferric transferrin over the course of the reaction occurs, until N-monoferric and apotransferrins are the only two species remaining. Essentially all of the apo form is from removal of iron from C-monoferric transferrin; iron is easily removed from both the N- and C-lobes in the diferric form, but there is no significant rate of iron removal from N-monoferric transferrin. Therefore, the vacancy of the C-lobe exerts a dramatic negative cooperativity upon iron removal from the N-lobe. Iron removal using every ligand shown in Fig. 2 also exhibited a similar buildup of N-monoferric transferrin. Since these ligands differ significantly (different chelating groups, denticities, iron affinities, etc.), we conclude that this effect is general for at least most ligands under physiological conditions.

Iron removal with TREN-3,2-HOPO as studied by gel electrophoresis. The first column is at 5 min, with subsequent 5 min intervals
Temperature, NaCl concentrations, and NaHCO3 concentrations were varied in order to determine if these were responsible for the observed sluggish removal of iron from the N-monoferric transferrin. Lowering the temperature to 30 or 25 °C slowed the rate of iron removal overall, but N-monoferric transferrin accumulation was still observed. Changing the bicarbonate concentration had little effect on the rate of iron removal, and N-monoferric transferrin accumulated. However, changing the chloride ion concentration affected both the overall rate of iron removal and the degree of N-monoferric transferrin accumulation. Increasing chloride ion concentration from 50 mM to 300 mM increased the overall rate of iron removal and changed the kinetic behavior of N-monoferric transferrin. At 50, 100, and 300 mM NaCl, no N-monoferric transferrin accumulation is observed (Fig. 4). Therefore, large cooperativity appears only with 150 mM of added NaCl, near the physiological chloride ion concentration.

Effect of chloride ion concentration on cooperativity. Each first column is at 10 min, with subsequent 10 min intervals (start and interval are 3 min for the 300 mM runs). The chelator used was TREN-3,2-HOPO
The effect of pH on the cooperativity was also studied. Figure 5 shows the gels for iron release in the pH range 7.4–6.3 with and without added ligand. As expected, iron release is more facile at lower pH values. At pH 6.3, the reaction rate is too fast to observe using gel electrophoresis. A comparison of the results between pH 6.5 and 7.4 illustrates that the accumulation of N-monoferric transferrin increases with increasing pH. This accumulation must result from either a decrease in the relative rate of iron removal from N-monoferric transferrin (decreasing k2N) or an increase in the relative rate of iron removal from the C-lobe in diferric transferrin (increasing k1C). These two possibilities can be distinguished by considering the effect on the diferric and apo forms of transferrin. The gels in Fig. 6 clearly show that iron release is facilitated by low pH; therefore, the buildup of N-monoferric transferrin at higher pH results from a decrease of k2N. This shows that cooperativity between the two lobes decreases as the pH is lowered.

Effect of pH on cooperativity. The first column is at 5 min, with subsequent 5 min intervals. The chelator used was TREN-3,2-HOPO. At each pH the first three lanes are without chelator, and the second three are with chelator

Proposed mechanism for iron removal. FeN refers to iron in the N-lobe. FeC refers to iron in the C-lobe. Left side of Tf represents the N-lobe. Right side of Tf represents the C-lobe. The asterisk refers to an “open” conformation of transferrin
Discussion
Despite a great deal of effort, the detailed molecular steps in iron removal from transferrin have remained rather poorly understood. Iron removal from the protein clearly depends on several factors, and seemingly small variations in reaction conditions can often dramatically change the kinetic behavior of transferrin. As a further complication, the literature contains some apparently contradictory results that are difficult to interpret [15, 20, 22, 25, 26, 30, 35, 36, 39, 41, 42, 43, 44, 47, 48, 50, 51, 57, 58, 59, 60, 61, 63, 64, 67, 80]. A general mechanism that is consistent with all the available data has remained an elusive goal.
Our results demonstrate large cooperativity between the two lobes, irrespective of the chelating ligand used at chloride ion concentrations near physiological conditions. We propose the mechanism shown schematically in Fig. 6 to explain the cooperativity. The salient feature of this mechanism is that iron removal from the N-lobe requires closure of the C-lobe. In the diferric form of transferrin, both lobes are closed and so anion-triggered opening of either site makes iron removal facile from either site. Similarly, iron removal from the C-monoferric form of transferrin can be triggered by anion binding; however, for the N-monoferric form of transferrin the C-lobe is vacant and open, blocking the anion binding to the N-lobe and making iron removal slow. Thus conditions that greatly decrease the amount of this form of transferrin should show accumulation of N-monoferric transferrin, as observed. This mechanism adequately explains our results concerning cooperativity; however, of greater interest is whether this mechanism can also be extended to other studies and transferrin iron removal in general. Examination of several iron removal studies suggests that indeed this is the case, and many of the apparent contradictions in the literature can be explained.
There is general agreement that an important early step in the removal of iron is a conformational change from the resting “closed” ferric lobe to an accessible “open” ferric lobe. It is also worth noting that X-ray solution scattering as well as X-ray crystallography has proven that there is a large conformational change from the “closed” ferric form to an “open” apo form [17, 18, 81, 82, 83, 84]. This mechanism, first proposed by Bates and co-workers [85] and modified by Chasteen and colleagues [34], accounts for saturation behavior with respect to increasing ligand concentration that is often (but not always) observed for iron removal.
There is also general agreement that transferrin must bind anions in order to release iron (an anion-binding site was identified for the C-terminal lobe, although this study was carried out at pH 5.6 [38]). Iron release occurs very slowly in the absence of added anions [38]; the rates of iron removal by a wide variety of ligands extrapolate to zero at zero ionic strength [59]. It has therefore been proposed that at least one kinetically significant anion-binding site must be occupied by an anion for iron removal, and this anion binding induces a conformational change from “closed” to “open”.
While the Bates mechanism explains the general features of the iron removal process, it must be incomplete. The two lobes are not kinetically equivalent and often display quite different responses to different conditions. Indeed, the relative lability of the two sites can be reversed with the appropriate choice of conditions [20, 22, 42, 44, 58, 59, 60, 64]. The mechanism also does not explain the effect of anions on iron release. Anions have been reported to accelerate iron removal from both lobes [24, 35, 36, 38, 42, 58, 59, 60, 63, 64, 80] and to accelerate iron removal from the C-lobe, while slowing (or having no effect on) iron removal from the N-lobe [20, 42, 44, 60]. Although the Bates model predicts that the removal of iron from transferrin should always produce saturation kinetics with respect to ligand at sufficiently high ligand concentrations, some studies have shown iron removal to exhibit a simple linear dependence on ligand concentration [26, 43] and others exhibit biphasic behavior (saturation kinetics followed by linear ligand dependence) [23, 26, 27, 30, 35, 43, 44, 64]. The Bates model also predicts that the maximum rate of iron removal should be the same for all ligands (because at saturation the rate-determining step is the conformational change of the protein), but this is not the case [41]. Finally, the Bates model includes no cooperativity between the two lobes, although small cooperativity has been reported several times [26, 27, 44, 58] and this work and two other studies have shown that substantial cooperativity also exists under certain conditions [14, 21]. A general mechanism must, therefore, address the following: differences between the kinetic iron removal behavior of the N- and C-lobe, variable ligand concentration dependence, anion effects, and cooperativity.
We compare the rates of iron removal from monoferric transferrins and a variety of ligands in Table 1. We reviewed an earlier study of iron removal rates from both monoferric transferrins with 3,4-LICAMS at 25 °C and 37 °C with and without added chloride ion (Table 2) [59]. Adding chloride ion and/or increasing the temperature increased both k2N and k2C, with the addition of chloride ion having a larger effect on the iron removal rates than the temperature increase. Interestingly, the ratio of k2N to k2C decreases when chloride ion is added or the temperature is increased, with k2N becoming smaller than k2C at 37 °C and 0.5 M chloride ion concentration [59]. The ratio of acceleration due to added chloride ion at 25 °C versus 37 °C [from Table 2; (A/B)/(C/D)] is 1.0 for monoferric C (k2C) and 1.4 for monoferric N (k2N). This means that the acceleration due to adding chloride ion and increasing temperature is additive for the C-lobe, but there is antagonism between them in the N-lobe. This antagonism is explained if opening of the C-lobe impedes opening of the N-lobe. Furthermore, the seemingly contradictory behavior of anions on iron removal from N-monoferric transferrin are explained, since factors that promote lobe opening could either promote N-lobe opening or hinder C-lobe closing. It also explains why large cooperativity has only rarely been observed, since the balance of factors generating this behavior occurs only near physiological chloride ion concentration. Cooperativity between the two lobes is very sensitive to the conditions employed, since iron removal from the N-lobe requires the C-lobe to be closed, which is more difficult if the C-lobe is vacant.
The iron-binding pockets of the two lobes in the protein are far apart from each other (>40 Å) [86, 87]; thus it is difficult to imagine a way in which opening of one lobe could directly block access to the other lobe. We suggest instead that the “open” C-lobe conformation blocks access to the anion-binding site in the N-lobe, which must be occupied before the “open” N-lobe conformation can form. This provides a good explanation for large cooperativity, since the anion-binding site need not be so close to the iron-binding region (and thus potentially much closer to the other lobe) as well as helping to explain the chloride ion dependence on cooperativity.
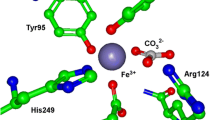
Support for this suggestion comes from study of recombinant N-lobe-only transferrin. This monolobal transferrin simplifies the problem by eliminating lobe–lobe interactions and allows facile site-directed mutagenesis [48, 49, 50, 51, 52, 53, 61, 88, 89, 90, 91]. It was assumed that lobe–lobe interactions are unimportant and that the isolated N-lobe behaves analogously to the N-lobe in holotransferrin. The present study shows the former to be incorrect, since large cooperativity must result from very significant lobe–lobe interactions. To check the latter assumption, we compared our data (the removal of iron from diferric transferrin by 300 equivalents of tiron at 0.14 mM chloride ion concentration and 25 °C) to those reported for the isolated N-lobe under the same conditions. The half-life reported for this reaction was 42 min with the isolated N-lobe, while all the iron from both lobes was removed in our study after only 5 min (gel electrophoresis). Assuming that the complete removal of iron observed represents at least 10 half-lives, the half-life for the holotransferrin is no more than 30 s (an increase of at least two orders of magnitude relative to the isolated N-lobe protein). [Thus isolated N-lobe monoferric transferrin does not model holotransferrin well because it has no interaction C-lobe connected to it.] We propose that the anion-binding site must at least partially reside on the linker or on the C-lobe. This explanation is attractive since it explains the different behavior of the isolated N-lobe and why conformational changes in the C-lobe have such a dramatic effect on iron removal from the N-lobe. Several studies have unsuccessfully attempted to locate the anion-binding site [39, 40, 45, 46, 48, 49, 50, 51, 52, 53, 62, 65, 84]; attention has been directed to residues located away from the lobe junction, since there are several positively charged residues in this area that could bind an anion. Figure 7 shows a pictorial representation of how we propose an “open” C-lobe is able to block the anion-binding site of the N-lobe, which prevents opening of the N-lobe.

Pictorial representation of the anion-binding site cooperativity and proposed mechanism. The diferric form of the protein (top) has both lobes in the closed conformation. Anion binding at the C-terminal site triggers opening of the protein and loss of iron to an external chelator. However, this conformational change blocks the anion-binding site of the N terminal lobe and hence its ability to release Fe(III)
The conformational change between the “closed” and “open” forms of a ferric lobe requires specific binding of an anion bound at the anion-binding site. Assuming the anion-binding constants are not radically different from each other (and the ability of the different anions to induce an “open” conformation is roughly the same), then concentrations of these anions should be the most important factor in determining which anion induces formation of the “open” conformation. For an efficient chelating ligand (only 10–20 equivalents necessary for fast iron removal), saturation behavior is seen, since most of the anions in solution are either from added salt or the buffer. Since less efficient chelators require higher concentrations, the added ligand affects the conformational change of the protein. In this case, small differences in anion concentrations, anion-binding constants, and the anion’s ability to promote formation of an “open” conformation mask the cooperativity. We suggest that this explains the often complicated behavior reported, particularly for iron removal from the N-lobe [20, 35, 36, 42, 60, 63, 64].
Conclusions
It is evident from the gel electrophoresis study that large cooperativity between transferrin’s two iron-binding sites is strong at 37 °C and 150 mM chloride ion concentration. Since this simulates physiological conditions, this work has major implications for the functional behavior of transferrin. It is known that the dominant form of transferrin in the body is the N-monoferric form, despite the fact that this is the less thermodynamically stable site [92]; perhaps the large cooperativity that we have observed explains the predominance of the N-monoferric transferrin. Also significant is the fact that this cooperativity vanishes as the pH is lowered from 7.4. Thus iron release in the endosome (pH 5.5) would not exhibit cooperativity, and both iron atoms are easily removed. It is known that diferric transferrin has a much higher binding affinity for the transferrin receptor than either monoferric form at pH 7.4 [17, 19]. Perhaps occupation of the C-lobe, which is significant only for the diferric form, is the trigger for receptor binding and iron delivery to cells, while the N-lobe serves in an iron sequestering role. It has been shown that certain bacteria contain a C-lobe receptor, which allow the bacteria to internalize ferric transferrin [93], and we have shown that siderophores have difficulty removing iron from N-monoferric transferrin [94]. Therefore, the large cooperativity might function to inhibit bacterial iron acquisition at pH 7.4 while retaining the ability to deliver iron to mammalian cells. Thus, the evolutionary advantage conferred by the genetic duplication of the monolobal transferrin progenitor might have been an early step in the iron acquisition “arms race” between multicellular organisms and their bacterial parasites.
References
Graham G, Bates GW, Rachmilewitz EA, Hershko C (1979) Am J Hematol 6:207–217
Chasteen ND, Woodworth RC (1990) Iron transport and storage. CRC Press, Boca Raton, Fla., USA
Agarwal MB, Gupte SS, Viswanathan C, Vasandani D, Ramanathan J, Desai N, Puniyani RR (1992) Br J Haematol 82:460–466
Al-Refaie FN, Hershko C, Hoffbrand AV, Kosaryan M, Olivieri NF, Tondury P, Wonke B (1995) Br J Haematol 91:224–229
Baldwin GS (1993) Comp Biochem Physiol B 106:203
Bartfeld NS, Law JH (1990) J Biol Chem 265:21684–21691
Gasdaska JR, Law JH, Bender CJ, Aisen P (1996) J Inorg Biochem 64:247–258
Park I, Schaeffer E, Sidoli A, Baralle FE, Cohen GN, Zakin MM (1985) Proc Natl Acad Sci USA 82:3149–3153
Williams J, Grace SA, Williams JM (1982) Biochem J 201:417–419
Huebers HA, Huebers E, Finch CA, Martin AW (1982) J Comp Physiol 148:101–109
Lee MY, Huebers H, Martin AW, Finch CA (1978) J Comp Physiol 127:349–354
Jamroz RC, Gasdaska JR, Bradfield JY, Law JH (1993) Proc Natl Acad Sci USA 90:1320–1324
Kurama T, Kurata S, Natori S (1995) Eur J Biochem 228:229–235
Stinzi A, Raymond KN (2000) J Biol Inorg Chem 5:57–66
Turcot I, Stinzi A, Xu J, Raymond KN (2000) J Biol Inorg Chem 5:634–
Makey DG, Seal US (1976) Biochim Biophys Acta 453:250–256
Aisen P (1999) Metal Ions Biol Syst 36:585–631
Sun H, Li H, Sadler PJ (1999) Chem Rev 99:2817–2842
Richardson DR, Ponka P (1997) Biochim Biophys Acta 1331:1–40
Baldwin DA, de Sousa DMR (1981) Biochem Biophys Res Commun 99:1101–1107
Ford S, Cooper RA, Evans RW, Hider RC, Williams PH (1988) Eur J Biochem 178:477–481
Bali PK, Harris WR (1990) Arch Biochem Biophys 281:251–256
Bailey CT, Byrne C, Chrispell K, Molkenbur C, Sackett M, Reid K, McCollum K, Vibbard D, Catelli R (1997) Biochemistry 36:10105–10108
Baldwin DA (1980) Biochim Biophys Acta 623:183–198
Baldwin DA, Egan TJ, Marques HM (1990) Biochim Biophys Acta 1038:1–9
Bali PK, Harris WR, Nesset-Tollefson D (1991) Inorg Chem 30:502–508
Bali PK, Harris WR (1989) J Am Chem Soc 111:4457–4461
Bali PK, Aisen P (1992) Biochemistry 31:3963–3967
Bali PK, Aisen P (1991) Biochemistry 30:9947–9952
Bertini I, Hirose J, Luchinat C, Messori L, Piccioli M, Scozzafava A (1988) Inorg Chem 27:2405–2409
Carrano CJ, Raymond KN (1979) J Am Chem Soc 101:5401–5404
Castellano AC, Barteri M, Castagnola M, Bianconi A, Borghi E, Longa SD (1994) Biochem Biophys Res Commun 198:646–652
Chahine J-MEH, Pakdaman R (1995) Eur J Biochem 230:1102–1110
Cowart RE, Swope S, Loh TT, Chasteen ND, Bates GW (1986) J Biol Chem 261:4607–4614
Egan TJ, Ross DC, Purves LR, Adams PA (1992) Inorg Chem 31:1994–1998
Egan TJ, Zak O, Aisen P (1993) Biochemistry 32:8162–8167
Faller B, Nick H (1994) J Am Chem Soc 116:3860–3865
Foley AA, Bates GW (1988) Biochim Biophys Acta 965:154–162
Grady JK, Mason AB, Woodworth RC, Chasteen ND (1995) Biochem J 309:403–410
Grossman JG, Mason AB, Woodworth RC, Neu M, Lindley PF, Hasnain SS (1993) J Mol Biol 231:554–558
Harris WR (1984) J Inorg Biochem 21:263–276
Harris WR, Bali PK (1988) Inorg Chem 27:2687–2691
Harris WR, Bali PK, Crowley MM (1992) Inorg Chem 31:2700–2705
Harris WR, Bao G (1997) Polyhedron 16:1069–1079
Harris WR, Cafferty AM, Abdollahi S, Trankler K (1998) Biochim Biophys Acta 1383:197–210
Harris WR, Nesset-Tollefson D, Stenbach JZ, Mohammed-Hani N (1990) J Inorg Biochem 38:175–183
Harris WR, Rezvani AB, Bali PK (1987) Inorg Chem 26:2711–2716
He Q-Y, Mason AB, Nguyen V, MacGillivray RTA, Woodworth RC (2000) Biochem J 350:909–915
He Q-Y, Mason AB, Pakdaman R, Chasteen ND, Dixon BK, Tam BM, Nguyen V, MacGillivray RTA, Woodworth RC (2000) Biochemistry 39:1205–1210
He Q-Y, Mason AB, Tam BM, MacGillivray RTA, Woodworth RC (1999) Biochemistry 38:9704–9711
He Q-Y, Mason AB, Woodworth RC (1997) Biochem J 328:439–445
He Q-Y, Mason AB, Woodworth RC, Tam BM, MacGillivray RTA, Grady JK, Chasteen ND (1998) J Biol Chem 273:17018–17024
He Q-Y, Mason AB, Woodworth RC, Tam BM, Wadsworth T, MacGillivray RTA (1997) Biochemistry 36:5522–5528
Katoh A, Hida Y, Kamitani J, Ohkanda J (1998) J Chem Soc Dalton Trans 3859–3864
Kojima N, Bates GW (1979) J Biol Chem 254:8847–8854
Konopka K, Bindereif A, Neilands JB (1982) Biochemistry 21:6503–6508
Kontoghiorghes GJ (1995) Analyst 120:845–851
Kretchmar SA, Raymond KN (1986) J Am Chem Soc 108:6212
Kretchmar SA, Raymond KN (1988) Inorg Chem 27:1436–1441
Li Y, Harris WR (1998) Biochim Biophys Acta 1387:89–102
Li Y, Harris WR, Maxwell A, MacGillivray RTA, Brown T (1998) Biochemistry 37:14157–14166
MacGillivray RTA, Bewley MC, Smith CA, He Q-Y, Mason AB, Woodworth RC, Baker EN (2000) Biochemistry 39:1211–1216
Marques HM, Walton T, Egan TJ (1995) J Inorg Biochem 57:11–21
Marques HM, Watson DL, Egan TJ (1991) Inorg Chem 30:3758–3762
Mason AB, He Q-Y, Tam BM, MacGillivray RTA, Woodworth RC (1998) Biochem J 330: 35–40
Mecklenburg SL, Donohoe RJ, Olah GA (1997) J Mol Biol 270:739–750
Muralidhara BK, Hirose M (2000) J Biol Chem 275:12463–12469
Muralidhara BK, Hirose M (2000) Protein Sci 9:1567–1575
Nguyen SAK, Craig A, Raymond KN (1993) J Am Chem Soc 115:6758–6764
Oe H, Takahashi N, Doi E, Hirose M (1989) J Biochem 106:858–863
Ohkanda J, Kamitani J, Tokumitsu T, Hida Y, Konakahara T, Katoh A (1997) J Org Chem 62:3618–3624
Ohkanda J, Katoh A (1995) Tetrahedron 51:12995–13002
Ohkanda J, Katoh A (1996) Chem Lett 423–424
Okada S, Rossmann MD, Brown EB (1978) Biochim Biophys Acta 543:72–81
Pollack S, Vanderhoff G, Lasky F (1977) Biochim Biophys Acta 497:481–487
Rodgers SJ, Raymond KN (1983) J Med Chem 26:439–442
Scarrow RC, White DL, Raymond KN (1985) J Am Chem Soc 107:6540–6546
Stefanini S, Chiancone E, Cavallo S, Saez V, Hall AD, Hider RC (1991) J Inorg Biochem 44:27–37
Steinlein LM, Ligman CM, Kessler S, Ikeda RA (1998) Biochemistry 37:13696–13703
Zak O, Tam B, MacGillivray RTA, Aisen P (1997) Biochemistry 36:11036–11043
Jeffrey PD, Bewley MC, MacGillivray RTA, Mason AB, Woodworth RC, Baker EN (1998) Biochemistry 37:13978–13986
Grossman JG, Crawley JB, Strange RW, Patel KJ, Murphy LM, Neu M, Evans RW, Hasnain SS (1998) J Mol Biol 279:461–472
Mizutani K, Yamashita H, Kurokawa H, Mikami B, Hirose M (1999) J Biol Chem 274:10190–10194
MacGillivray RTA, Moore SA, Chen J, Anderson BF, Baker H, Luo Y, Bewley M, Smith CA, Murphy MEP, Wang Y, Mason AB, Woodworth RC, Brayer GD, Baker EN (1998) Biochemistry 37:7919–7928
Cowart RE, Kojima N, Bates GW (1982) J Biol Chem 257:7560–7565
Yajima H, Tetsuya S, Kikuchi T, Morita M, Ishii T (2000) J Protein Chem 19:215–223
Sun XL, Baker HM, Shrewry SC, Jameson GB, Baker EN (1999) Acta Crystallogr Sect D 55:403–XXX
He Q-Y, Mason AB, Woodworth RC, Tam BM, MacGillivray RTA, Grady JK, Chasteen ND (1997) Biochemistry 36:14853–14860
He Q-Y, Mason AB, Tam BM, MacGillivray RTA, Woodworth RC (1999) Biochem J 344:881–887
Steinlein LM, Graf TN, Ikeda RA (1995) Protein Express Purif 6:619–624
Mason AB, Woodworth RC, Oliver RWA, Green BN, Lin L-N, Brandts JF, Tam BM, Maxwell A, MacGillivray RTA (1996) Protein Express Purif 8:119–125
Leibman A, Aisen P (1979) Blood 53:1058–1065
Retzer MD, Kabani A, Button LL, Yu R-h, Schryvers AB (1996) J Biol Chem 271:1166–1173
Tillbrook GS (1999) Metal Ions Biol Syst 36:691–729
Acknowledgements
This research was supported by NIH grant DK 57814. We thank Emily Dertz for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hamilton, D.H., Turcot, I., Stintzi, A. et al. Large cooperativity in the removal of iron from transferrin at physiological temperature and chloride ion concentration. J Biol Inorg Chem 9, 936–944 (2004). https://doi.org/10.1007/s00775-004-0592-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-004-0592-6