
Abstract
Periodontitis, one of the most common infectious diseases in humans, is characterized by inflammation of the periodontal tissue and subsequent destruction of the alveolar bone, which ultimately leads to tooth loss. Recently, it was revealed that the osteoclastic bone damage that occurs during periodontitis is dependent on the receptor activator of NF-kB ligand (RANKL) produced by osteoblastic cells and periodontal ligament cells. Immune cells provide essential cues for the RANKL induction that takes place during periodontal inflammation. The knowledge accumulated and experimental tools established in the field of “osteoimmunology” have made crucial contributions to a better understanding of periodontitis pathogenesis and, reciprocally, the investigation of periodontitis has provided important insights into the field. This review discusses the molecular mechanisms underlying periodontal bone loss by focusing on the osteoimmune interactions and RANKL.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epithelial barrier function is essential for the maintenance of host-microbe homeostasis. The human body surface is mostly covered by an epithelial layer, a physical barrier functioning as the first line of defense against the invasion of pathogens as well as commensal microbes. The human oral barrier, which harbors around 700 bacterial species, is a unique exception as the integrity of the epithelium is physically breached by the teeth, and thus is susceptible to the infectious disease caused by oral bacteria, periodontitis [1, 2]. In severe periodontitis, oral bacteria and their components flow into the bloodstream via the inflamed gingiva and translocate to other organs [1, 2]. Thus, periodontitis may not only cause tooth loss, but also can affect systemic health. Epidemiological studies have suggested that periodontitis is associated with various systemic conditions, such as diabetes, cardiovascular diseases, adverse pregnancy outcomes, Alzheimer's disease and rheumatoid arthritis [1, 2].
RANKL (encoded by the Tnfsf11 gene) is the master regulator of osteoclast differentiation and function [1]. The indispensable role of RANKL in osteoclastogenesis was demonstrated by the finding that RANKL-deficient mice as well as RANKL-mutated patients exhibit osteopetrosis due to a complete lack of osteoclasts [3, 4]. It was also shown that RANKL-deficient mice were protected from bone erosion during arthritis, indicating that osteoclast formation under inflammatory conditions is also dependent on RANKL [5,6,7]. Thus, an understanding of the mechanisms underlying RANKL induction in periodontal lesions is vitally important for developing future therapeutic strategies for periodontal bone and tooth loss.
During periodontitis, immune cells accumulate in the periodontal lesions and induce local inflammation and osteoclastic bone damage. In 1972, Horton et al. reported that immune cells stimulated by the dental plaque derived from periodontitis patients produce osteoclast-activating factors, providing the first evidence for an interaction between immune and bone cells [8]. Subsequently, Takayanagi et al. explored the role of immune cells in bone damage in the context of rheumatoid arthritis, and showed that T cells can regulate osteoclastogenesis in a manner dependent on the balance between and among the cytokines that T cells produce [9]. This study explicitly illustrated the molecular mechanisms underlying the crosstalk between immune and skeletal systems, and the term “osteoimmunology” was coined by Arron and Choi to describe this interdisciplinary research in 2000 [10]. The last two decades have witnessed a critical contribution of osteoimmunological research to advances in the understanding the pathogenesis of and development of therapeutic strategies toward bone destruction in rheumatoid arthritis [1]. However, although the pioneering work in this field was conducted in the context of periodontitis [8], the mechanisms and the role of osteoimmune crosstalk in periodontitis were largely unclear until fairly recently.
The molecular pathogenesis of periodontal inflammation
Periodontitis had long been considered to be an infectious disease caused by pathogens such as Porphyromonas gingivalis (P.g.) based on the strong association of such pathogens with the disease sites. However, recent studies have indicated that periodontitis may not result from individual pathogens per se, rather from a dysbiosis of the oral commensal microbiome, which perturbs host-microbe homeostasis at the oral barrier [11]. Importantly, P.g. was shown to induce periodontitis in specific-pathogen-free (SPF) mice but not in germ-free (GF) mice, highlighting the essential role of the commensal microbiome in the pathogenesis of periodontitis [12, 13]. Furthermore, it was shown that an accumulation of oral commensal bacteria induced by the placement of a silk ligature around the tooth can cause severe periodontitis without P.g. infection in SPF mice [14]. These findings suggest that the alteration in the quality and quantity of the oral commensal bacteria is critical for the development of periodontitis.
Dysbiosis in the oral microbial community triggers a host immune response at the oral barrier [15, 16]. It is well established that CD4+ T cells play a central role in the immune system. The importance of CD4+ T cells in periodontitis has been evidenced in a number of studies showing that animals lacking CD4+ T cells are protected from periodontal inflammation and bone destruction [1]. Early studies proposed that Th1 cells may contribute to the tissue damage at an initial stage of periodontitis, whereas Th2 cells may be involved in disease progression [15]. However, Th1 cells and Th2 cells potently inhibit osteoclast differentiation via the secretion of IFN-γ and IL-4, respectively [4]. Therefore, from the osteoimmunological standpoint, it is difficult to construe periodontal bone damage as a consequence of the activation of Th1 or Th2 cells. Th17 cells and regulatory T (Treg) cells are newly identified CD4+ T cell subsets. Th17 cells play a key role in the host defense against extracellular bacteria and fungi by inducing the production of anti-microbial peptides by epithelial cells, neutrophil recruitment and local inflammation. Among CD4+ T cell subsets, Th17 cells have been shown to be the exclusive bone-damaging T cells capable of promoting osteoclast differentiation [4]. Treg cells suppress excessive and/or prolonged immune responses, and also potently inhibit osteoclastogenesis [4]. A previous report showed that the induction of Treg accumulation in the periodontal tissue can inhibit periodontitis in mice and canines [17], suggesting that Treg cells have a capacity to prevent periodontal inflammation but the function of Treg cells might be suppressed during periodontitis. Intriguingly, a certain population of Treg cells was shown to convert into Th17 cells under inflammatory conditions [18]. Th17 cells converted form Treg cells (called exFoxp3Th17 cells) have a much stronger pro-osteoclastogenic activity than conventional Th17 cells, and critically contribute to the bone destruction associated with arthritis [18].
Recent studies showed that Th17 cells and exFoxp3Th17 cells markedly accumulate in periodontal lesions [2, 19]. The accumulation of these cells was shown to be dependent on the oral commensal bacteria and the IL-6 produced by periodontal stromal cells [2, 19]. Mouse models that lack Th17 cells (Stat3flox/flox CD4-Cre mice and Rorcflox/flox Lck-Cre mice), exFoxp3Th17 cells (Il6raflox/flox Foxp3-Cre mice) and IL-17A/F (Il17a–/–Il17f–/– mice) developed less inflammation and bone destruction during the course of periodontitis [2, 19]. In human patients, IL-17+CD4+ T cells were found to be expanded in the periodontal lesions, and STAT3-mutated patients with genetic defects in Th17 cell differentiation were protected from periodontal inflammation and bone loss [19]. It was also reported that Foxp3+IL-17+ T cells, which are considered to be in a transition state during the conversion from Treg cells into exFoxp3Th17 cells, are frequently observed in the periodontal lesions of patients with severe periodontitis [20]. Antibiotic treatment was shown to decrease the frequency of Foxp3+IL-17+ T cells in periodontitis patients [21]. In patients having both rheumatoid arthritis and periodontitis, blockade of IL-6R by Tocilizumab was shown to significantly inhibit periodontal inflammation [22]. Leukocyte adhesion deficiency type 1 (LAD1) patients develop severe periodontitis caused by an abnormal expansion of Th17 cells in the periodontal tissues, leading to a complete loss of adult teeth in most of the patients [23]. Ustekinumab, an antibody that binds to the p40 subunit of IL-23 and IL-12, has been shown to ameliorate periodontitis in LAD1 patients by inhibiting the activation of Th17 cells in the oral mucosa [23]. These findings suggest that Th17 cells and exFoxp3Th17 cells play a central role in periodontal inflammation.
In addition to CD4+ T cells, various types of other immune cells are involved in the pathogenesis of periodontitis. Neutrophils are antimicrobial effector cells and abundantly present in periodontal lesions. The precise coordination of neutrophil recruitment is essential for periodontal tissue homeostasis, as evidenced by findings that either too few or too many neutrophils exacerbate periodontitis in mice and humans [16]. A previous report showed that bone loss was partly inhibited in B cell-deficient mice in the late phase, but not in the early phase of a murine periodontitis model, suggesting that B cells contribute to tissue damage in chronic periodontitis [24]. Antigen presenting cells, including dendritic cells and macrophages, support the development of Th17 cells in periodontal lesions, and may also contribute to inflammation and bone destruction by producing inflammatory cytokines such as IL-1β [1].
Immune cells are also involved in the resolution of inflammation. Th17 cells were shown to transdifferentiate into Treg cells during the resolution phase of colitis, helminth infection and multiple sclerosis [25]. Although it is still unknown if the conversion from Th17 cells into Treg cells contributes to the resolution of periodontal inflammation, the plasticity of CD4+ T cells is likely to be involved in both the exacerbation and remission of periodontitis. Developmental endothelial locus-1 (DEL-1), an endothelial cell-secreted protein inhibiting neutrophil recruitment, is a key osteoimmune factor that suppresses both inflammation and osteoclastogenesis [26]. Macrophages were shown to produce DEL-1 and thus to promote the resolution of periodontitis by facilitating “efferocytosis”, a macrophage-mediated clearance of apoptotic neutrophils [27]. Langerhans cells were reported to inhibit periodontal inflammation by increasing the Treg cell number in periodontal tissues via unknown mechanisms [28]. Gingival γδT cells were also shown to promote the healing of periodontal lesions by producing amphiregulin [29]. Epithelial cells and plasma cells express IL-37 in human periodontal tissue [30]. Specific variants of the IL-37 gene locus with decreased IL-37 expression were shown to be associated with high IL-1β levels in the gingival crevicular fluid and the severity of periodontitis, suggesting that IL-37 has an inhibitory effect on periodontal inflammation [30].
These findings highlight the importance and complexity of the immune cell network in the pathogenesis of periodontitis. Further studies are needed to gain a comprehensive picture of how immune cells contribute to the physiology and pathology of the oral barrier system.
The cellular sources of RANKL and the mechanisms underlying periodontal bone loss
Since RANKL is the master regulator of osteoclastogenesis and play an essential role in osteoclast-associated diseases, researchers in the field of periodontology have long explored the cellular source of RANKL and the mechanisms underlying RANKL induction during periodontitis [31, 32]. The levels of RANKL mRNA in the periodontal tissue and RANKL protein in the gingival crevicular fluid are associated with the severity of periodontitis in patients [31, 32]. Among the cells that constitute the periodontal tissue, osteoblasts, osteocytes, periodontal ligament cells and gingival epithelial cells have been shown to have the capacity to support osteoclastogenesis by producing RANKL in in vitro co-culture systems [33,34,35,36]. Previous reports of studies using immunohistochemical analyses with anti-RANKL antibodies proposed that RANKL is expressed in B cells, T cells, fibroblasts and epithelial cells in periodontal tissues [37, 38]. However, the expression level of the RANKL protein is very low even in osteoblastic cells, and the detection of RANKL by immunohistochemistry is technically difficult. In situ hybridization experiments showed that RANKL mRNA is expressed in mesenchymal cells adjacent to the alveolar bone during periodontitis [2] (Fig. 1).

Histological analysis of RANKL expression in the periodontal lesion. In situ hybridization of Tnfsf11 mRNA in the periodontal tissues of control mice or periodontitis-induced wild-type mice (5 days after the ligature placement). The black arrows indicate osteoclasts (multinucleated giant cells) and the black arrowheads indicate RANKL-expressing mesenchymal cells, including osteoblastic cells (cuboidal cells on the alveolar bone surface) and periodontal ligament fibroblasts (spindle-shape cells connecting teeth to the alveolar bone). H&E haematoxylin and eosin stain, T tooth; AB alveolar bone. A representative image of more than three independent experiments is shown
RANKL is synthesized in a membrane-bound form, and cleaved into its soluble form by proteases. Although both forms can function as agonistic ligands for the receptor RANK, RANKL is considered to be mainly functional in its membrane-bound form [4]. Importantly, physiological bone remodeling was normal in Tnfsf11ΔS/ΔS mice, in which the soluble form of RANKL is absent and only membrane-bound RANKL exists, indicating that soluble RANKL is dispensable for bone homeostasis at steady state [39]. However, certain reports have proposed that soluble RANKL plays a major role in periodontal bone loss based on in vitro experiments [37, 40]. It is important to know which form of RANKL contributes to periodontal bone loss to understand the anatomical localization of cellular sources of RANKL in periodontitis. For instance, if the membrane-bound form plays a major role, RANKL-producing cells should be localized in close proximity to the alveolar bone, and the contribution of RANKL expressed in cells that do not come into contact with bone, such as epithelial cells and most hematopoietic cells, may be ruled out. Indeed, it was shown that there is no difference in periodontitis-induced bone loss between Tnfsf11ΔS/ΔS mice and wild-type mice, indicating that the contribution of soluble RANKL is negligible in periodontal bone loss [2]. Therefore, the cellular sources of RANKL are suggested to be in direct contact with the alveolar bone.
To identify the major sources of RANKL in periodontitis, RANKL-floxed mice were crossed with various Cre lines specific to B cells (Mb1-Cre), T cells (CD4-Cre), osteoblastic cells (Sp7-tTA-tetO-Cre) and periodontal ligament cells (Scleraxis-Cre) [2]. An inducible Cre system (Sp7-tTA-tetO-Cre) was used for deleting RANKL in osteoblastic cells, because the RANKL on osteoblastic cells is essential for tooth eruption [41]. In the Sp7-tTA-tetO-Cre system, Cre recombinase is expressed only when a tetracycline-controlled transactivator (tTA) binds to a tetracycline responsive element (tetO) in the absence of doxycycline (Dox) [2]. Sp7-tTA-tetO-Cre mice crossed with RANKL-floxed mice were treated from the prenatal period with Dox, which was withdrawn at the age of 3 weeks [2] (Fig. 2). The RANKL conditional knockout mice were subjected to the ligature-induced periodontitis at the age of 8 weeks and alveolar bone was analyzed 10 days after the ligature placement in all groups [2]. Periodontitis-induced bone loss and osteoclast development were markedly suppressed when RANKL was deleted in Sp7-Cre expressing osteoblastic cells and Scleraxis-Cre expressing periodontal ligament cells, indicating that these mesenchymal cells are the major source of RANKL in periodontitis [2] (Fig. 2). However, since periodontal ligament cells contain progenitor cells that give rise to osteoblastic cells [42], and Sp7+/Scleraxis+, Sp7+/Scleraxis– and Sp7–/Scleraxis+ cell populations are present in the periodontal tissue [43], it is difficult to rigorously discriminate between periodontal ligament and osteoblastic cells using the Sp7-Cre and Scleraxis-Cre systems. Further studies are required to clarify relative contribution of RANKL on periodontal ligament and osteoblastic cells to periodontal bone loss. Another study also showed that RANKL-floxed mice crossed with Dmp1-Cre mice, in which Cre recombinase is expressed in osteoblastic cells including osteoblasts and osteocytes, were protected from periodontitis-induced bone loss [44].

Osteoblastic cells and periodontal ligament cells are the major source of RANKL in periodontal bone loss. a Schematic of the experimental system for the ligature-induced periodontitis model in the RANKL conditional knockout mice [2]. b, c Periodontal bone loss evaluated by micro-CT analysis (b) and osteoclast number in the maxilla evaluated by histological analysis (c) in control mice (Tnfrsf11flox/flox) and RANKL conditional knockout mice in which RANKL was specifically deleted in B cells (Tnfrsf11flox/flox Mb1- Cre), T cells (Tnfrsf11flox/flox Cd4-Cre), periodontal ligament cells (Tnfrsf11flox/flox Scx-Cre) or osteoblastic cells (Tnfrsf11flox/flox Sp7-Cre) [2]. Statistical analyses were performed using ANOVA with Dunnett's multiple-comparison test. *P < 0.05; ***P < 0.005; N.S. not significant
Among the T-cell subsets, exFoxp3Th17 cells express RANKL most highly at both the mRNA and protein levels [2, 18]. exFoxp3Th17 cells highly express membrane-bound RANKL as well as adhesion molecules such as integrins, having a capacity to directly come in contact with osteoclast precursor cells [2, 18]. Periodontitis-induced bone loss and osteoclast formation were significantly reduced when RANKL was deleted in T cells, but not in B cells [2]. However, the effect of RANKL deletion in T cells was less than the deletion of RANKL in mesenchymal cells [2] (Fig. 2). Previous studies showed that IL-17 stimulates RANKL expression in osteoblastic cells and periodontal ligament cells [45, 46]. Thus, it is likely that Th17 cells and exFoxp3Th17 cells contribute to periodontal bone loss, mainly by inducing RANKL expression on mesenchymal cells via IL-17 production.
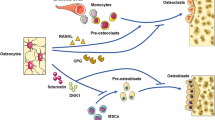
Osteoprotegerin (OPG), a decoy receptor for RANKL, is a key osteoprotective factor playing a central role in bone homeostasis. Oral bacteria- and osteoclast-derived proteases were shown to cleave OPG and promote osteoclastogenesis in vitro [47,48,49]. OPG-deficient mice spontaneously developed severe alveolar bone loss [50], suggesting that not only the up-regulation of RANKL, but also the down-regulation and/or degradation of OPG is involved in periodontal bone loss. OPG is reported to be highly expressed in osteocytes in periodontal tissues [50], but recent studies using OPG-floxed mice showed that osteoblasts, but not osteocytes, are the primary source of OPG in the long bones and vertebral bones [51, 52]. Further studies are needed to specify the cellular source of OPG in alveolar bone homeostasis. Immune cell-derived inflammatory cytokines such as IL-17, TNF and IL-1β, as well as bacterial components (e.g., lipopolysaccharides) were reported to up-regulate the RANKL/OPG ratio in osteoblastic cells and periodontal ligament cells [1], so these factors may critically contribute to bone damage by inducing osteoclast formation on the alveolar bone during the course of periodontitis (Fig. 3). These findings suggest that therapeutic approaches targeting immune cells and RANKL, together with appropriate infection control, may be a promising strategy to prevent bone damage in periodontitis.

Molecular mechanisms of bone destruction in periodontitis. Poor oral hygiene and P.g. (Porphyromonas gingivalis) induce dysbiosis in oral microbial communities. Oral bacteria systemically disseminate via the inflamed gingiva, adversely affecting various systemic conditions. Oral bacteria and their components (e.g., lipopolysaccharides: LPS) stimulate innate immune cells such as macrophages and dendritic cells as well as periodontal ligament cells. Periodontal ligament cells produce IL-6, which induces the accumulation of Th17 cells and exFoxp3Th17 cells in the oral mucosa. They in turn produce IL-17, which contributes to the eradication of oral bacteria by inducing antibacterial peptides and neutrophil chemo-attractants in the gingival epithelial cells. IL-17 also stimulates osteoclastic bone resorption by inducing receptor activator of nuclear factor-κB ligand (RANKL) in osteoblasts and periodontal ligament cells. Bacteria-derived proteases degrade osteoprotegerin (OPG). Other inflammatory cytokines such as the IL-1 and TNF produced by macrophages and dendritic cells as well as bacterial products also stimulate periodontal ligament cells and osteoblasts so as to increase the RANKL/OPG ratio, promoting osteoclastic bone erosion
The evolutionary origin of inflammatory bone destruction–a hypothesis
In rheumatoid arthritis, Th17-cell-mediated bone damage is exclusively pathogenic, but it may be related to the host defense machinery against bacterial infection in the context of periodontitis. During periodontitis, Th17 cells, including exFoxp3Th17 cells, eradicate oral bacteria in two different ways. First, they evoke anti-bacterial responses, inducing anti-bacterial peptides as well as neutrophil chemo-attractants in oral epithelial cells. Second, they stop prolonged infection and inflammation by promoting osteoclastic bone resorption, leading to the ejection of the diseased-tooth which is the source of infection (Fig. 3). Indeed, tooth loss completely terminates periodontitis-induced local inflammation and the systemic dissemination of oral bacteria [2]. Needless to say, the teeth are essential and non-substitutable organ for humans. Since tooth loss has adverse effects on human health, it is difficult to say whether the infection-driven tooth loss is ultimately “beneficial”. However, in the short run, it may well be. It is reported that osteopetrotic patients with severe periodontitis develop prolonged infection and life-threatening osteomyelitis of the jaw, which is treated by extraction of the diseased-tooth and antibiotic treatment [53]. Medication-related osteonecrosis of the jaw (MRONJ) is one of the most serious side effects of anti-bone resorptive drugs. Although tooth extraction has been considered a potential trigger for the onset of MRONJ, recent studies have suggested that local infection, but not tooth extraction itself, is the key risk factor [1]. From an evolutionary point of view, most of vertebrates are polyphyodonts, which means they can continually replace lost teeth. Infection-driven tooth loss is also observed in ancient reptiles, and the Tnfsf11 and Il17a genes are widely conserved across species ranging from jawed fishes to humans [54, 55]. These findings suggest that the bone loss mechanism exerted by Th17 cells may have emerged as a primitive host defense mechanism for stopping prolonged bacterial infection at the oral barrier, a system which has been evolutionarily conserved from polyphyodont animals with substitutable teeth [1, 55].
Concluding remarks
Periodontitis is a unique osteoimmune disease in which RANKL links the antibacterial immune response to bone destruction. There are few other conditions in which infection occurs around the bone, other than periodontitis. The field of osteoimmunology has developed and progressed with investigations into the mechanisms underlying bone damage associated with autoimmune arthritis, and substantially contributed to a better understanding of the pathogenesis of inflammatory bone diseases, including periodontitis. Since the primary role of the immune system is to provide host defense against pathogens, the investigation of periodontitis will help illuminate the “true nature” of the interactions between immune cells and bone cells, which cannot be understood by arthritis research alone. Periodontal research is now one of the critical domains of the field of osteoimmunology. Future investigations into the molecular mechanisms of periodontitis will provide key insights that will contribute to advances in therapeutic strategies not only for periodontitis, but also for various diseases in which bone and/or immune cells are involved.
References
Tsukasaki M, Takayanagi H (2019) Osteoimmunology: evolving concepts in bone-immune interactions in health and disease (in eng). Nat Rev Immunol 19:626–642. https://doi.org/10.1038/s41577-019-0178-8
Tsukasaki M, Komatsu N, Nagashima K, Nitta T, Pluemsakunthai W, Shukunami C, Iwakura Y, Nakashima T, Okamoto K, Takayanagi H (2018) Host defense against oral microbiota by bone-damaging T cells (in eng). Nat Commun 9:701. https://doi.org/10.1038/s41467-018-03147-6
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM (1999) OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis (in eng). Nature 397:315–323. https://doi.org/10.1038/16852
Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, Sawa S, Nitta T, Takayanagi H (2017) Osteoimmunology: the conceptual framework unifying the immune and skeletal systems (in eng). Physiol Rev 97:1295–1349. https://doi.org/10.1152/physrev.00036.2016
Pettit AR, Ji H, von Stechow D, Müller R, Goldring SR, Choi Y, Benoist C, Gravallese EM (2001) TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis (in eng). Am J Pathol 159:1689–1699. https://doi.org/10.1016/S0002-9440(10)63016-7
Tsukasaki M, Hamada K, Okamoto K, Nagashima K, Terashima A, Komatsu N, Win SJ, Okamura T, Nitta T, Yasuda H, Penninger JM, Takayanagi H (2017) LOX fails to substitute for RANKL in osteoclastogenesis (in eng). J Bone Miner Res 32:434–439. https://doi.org/10.1002/jbmr.2990
Tanaka S (2017) RANKL-independent osteoclastogenesis: a long-standing controversy (in eng). J Bone Miner Res 32:431–433. https://doi.org/10.1002/jbmr.3092
Horton JE, Raisz LG, Simmons HA, Oppenheim JJ, Mergenhagen SE (1972) Bone resorbing activity in supernatant fluid from cultured human peripheral blood leukocytes (in eng). Science 177:793–795
Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T (2000) T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma (in eng). Nature 408:600–605. https://doi.org/10.1038/35046102
Arron JR, Choi Y (2000) Bone versus immune system (in eng). Nature 408:535–536. https://doi.org/10.1038/35046196
Hajishengallis G (2015) Periodontitis: from microbial immune subversion to systemic inflammation (in eng). Nat Rev Immunol 15:30–44. https://doi.org/10.1038/nri3785
Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA (2011) Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement (in eng). Cell Host Microbe 10:497–506. https://doi.org/10.1016/j.chom.2011.10.006
Maekawa T, Krauss JL, Abe T, Jotwani R, Triantafilou M, Triantafilou K, Hashim A, Hoch S, Curtis MA, Nussbaum G, Lambris JD, Hajishengallis G (2014) Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis (in eng). Cell Host Microbe 15:768–778. https://doi.org/10.1016/j.chom.2014.05.012
Abe T, Hajishengallis G (2013) Optimization of the ligature-induced periodontitis model in mice (in eng). J Immunol Methods 394:49–54. https://doi.org/10.1016/j.jim.2013.05.002
Hajishengallis G, Korostoff JM (2017) Revisiting the page and Schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later (in eng). Periodontol 75:116–151. https://doi.org/10.1111/prd.12181
Moutsopoulos NM, Konkel JE (2018) Tissue-specific immunity at the oral mucosal barrier (in eng). Trends Immunol 39:276–287. https://doi.org/10.1016/j.it.2017.08.005
Glowacki AJ, Yoshizawa S, Jhunjhunwala S, Vieira AE, Garlet GP, Sfeir C, Little SR (2013) Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes (in eng). Proc Natl Acad Sci USA 110:18525–18530. https://doi.org/10.1073/pnas.1302829110
Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H (2014) Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis (in eng). Nat Med 20:62–68. https://doi.org/10.1038/nm.3432
Dutzan N, Kajikawa T, Abusleme L, Greenwell-Wild T, Zuazo CE, Ikeuchi T, Brenchley L, Abe T, Hurabielle C, Martin D, Morell RJ, Freeman AF, Lazarevic V, Trinchieri G, Diaz PI, Holland SM, Belkaid Y, Hajishengallis G, Moutsopoulos NM (2018) A dysbiotic microbiome triggers T (in eng). Sci Transl Med. https://doi.org/10.1126/scitranslmed.aat0797
Okui T, Aoki Y, Ito H, Honda T, Yamazaki K (2012) The presence of IL-17+/FOXP3+ double-positive cells in periodontitis (in eng). J Dent Res 91:574–579. https://doi.org/10.1177/0022034512446341
Rajendran M, Looney S, Singh N, Elashiry M, Meghil MM, El-Awady AR, Tawfik O, Susin C, Arce RM, Cutler CW (2019) Systemic antibiotic therapy reduces circulating inflammatory dendritic cells and Treg-Th17 plasticity in periodontitis (in eng). J Immunol 202:2690–2699. https://doi.org/10.4049/jimmunol.1900046
Kobayashi T, Okada M, Ito S, Kobayashi D, Ishida K, Kojima A, Narita I, Murasawa A, Yoshie H (2014) Assessment of interleukin-6 receptor inhibition therapy on periodontal condition in patients with rheumatoid arthritis and chronic periodontitis (in eng). J Periodontol 85:57–67. https://doi.org/10.1902/jop.2013.120696
Moutsopoulos NM, Zerbe CS, Wild T, Dutzan N, Brenchley L, DiPasquale G, Uzel G, Axelrod KC, Lisco A, Notarangelo LD, Hajishengallis G, Holland SM (2017) Interleukin-12 and interleukin-23 blockade in leukocyte adhesion deficiency type 1 (in eng). N Engl J Med 376:1141–1146. https://doi.org/10.1056/NEJMoa1612197
Abe T, AlSarhan M, Benakanakere MR, Maekawa T, Kinane DF, Cancro MP, Korostoff JM, Hajishengallis G (2015) The B cell-stimulatory cytokines BLyS and april are elevated in human periodontitis and are required for B cell-dependent bone loss in experimental murine periodontitis (in eng). J Immunol 195:1427–1435. https://doi.org/10.4049/jimmunol.1500496
Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H et al (2015) Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation (in eng). Nature 523:221–225. https://doi.org/10.1038/nature14452
Maekawa T, Hosur K, Abe T, Kantarci A, Ziogas A, Wang B, Van Dyke TE, Chavakis T, Hajishengallis G (2015) Antagonistic effects of IL-17 and D-resolvins on endothelial Del-1 expression through a GSK-3β-C/EBPβ pathway (in eng). Nat Commun 6:8272. https://doi.org/10.1038/ncomms9272
Kourtzelis I, Li X, Mitroulis I, Grosser D, Kajikawa T et al (2019) DEL-1 promotes macrophage efferocytosis and clearance of inflammation (in eng). Nat Immunol 20:40–49. https://doi.org/10.1038/s41590-018-0249-1
Arizon M, Nudel I, Segev H, Mizraji G, Elnekave M, Furmanov K, Eli-Berchoer L, Clausen BE, Shapira L, Wilensky A, Hovav AH (2012) Langerhans cells down-regulate inflammation-driven alveolar bone loss (in eng). Proc Natl Acad Sci USA 109:7043–7048. https://doi.org/10.1073/pnas.1116770109
Krishnan S, Prise IE, Wemyss K, Schenck LP, Bridgeman HM, McClure FA, Zangerle-Murray T, O’Boyle C, Barbera TA, Mahmood F, Bowdish DME, Zaiss DMW, Grainger JR, Konkel JE (2018) Amphiregulin-producing γδ T cells are vital for safeguarding oral barrier immune homeostasis (in eng). Proc Natl Acad Sci USA 115:10738–10743. https://doi.org/10.1073/pnas.1802320115
Offenbacher S, Jiao Y, Kim SJ, Marchesan J, Moss KL, Jing L, Divaris K, Bencharit S, Agler CS, Morelli T, Zhang S, Sun L, Seaman WT, Cowley D, Barros SP, Beck JD, Munz M, Schaefer AS, North KE (2018) GWAS for Interleukin-1β levels in gingival crevicular fluid identifies IL37 variants in periodontal inflammation (in eng). Nat Commun 9:3686. https://doi.org/10.1038/s41467-018-05940-9
Chen B, Wu W, Sun W, Zhang Q, Yan F, Xiao Y (2014) RANKL expression in periodontal disease: where does RANKL come from? (in eng). Biomed Res Int 2014:731039. https://doi.org/10.1155/2014/731039
Nagasawa T, Kiji M, Yashiro R, Hormdee D, Lu H, Kunze M, Suda T, Koshy G, Kobayashi H, Oda S, Nitta H (2000) Ishikawa I (2007) Roles of receptor activator of nuclear factor-kappaB ligand (RANKL) and osteoprotegerin in periodontal health and disease (in eng). Periodontol 43:65–84. https://doi.org/10.1111/j.1600-0757.2006.00185.x
Takahashi N, Akatsu T, Udagawa N, Sasaki T, Yamaguchi A, Moseley JM, Martin TJ, Suda T (1988) Osteoblastic cells are involved in osteoclast formation (in eng). Endocrinology 123:2600–2602. https://doi.org/10.1210/endo-123-5-2600
Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression (in eng). Nat Med 17:1231–1234. https://doi.org/10.1038/nm.2452
Kanzaki H, Chiba M, Shimizu Y, Mitani H (2002) Periodontal ligament cells under mechanical stress induce osteoclastogenesis by receptor activator of nuclear factor kappaB ligand up-regulation via prostaglandin E2 synthesis (in eng). J Bone Miner Res 17:210–220. https://doi.org/10.1359/jbmr.2002.17.2.210
Usui M, Sato T, Yamamoto G, Okamatsu Y, Hanatani T, Moritani Y, Sano K, Yamamoto M, Nakashima K (2016) Gingival epithelial cells support osteoclastogenesis by producing receptor activator of nuclear factor kappa B ligand via protein kinase A signaling (in eng). J Periodontal Res 51:462–470. https://doi.org/10.1111/jre.12323
Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, Goncalves RB, Valverde P, Dibart S, Li YP, Miranda LA, Ernst CW, Izumi Y, Taubman MA (2006) B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease (in eng). Am J Pathol 169:987–998. https://doi.org/10.2353/ajpath.2006.060180
Fujihara R, Usui M, Yamamoto G, Nishii K, Tsukamoto Y, Okamatsu Y, Sato T, Asou Y, Nakashima K, Yamamoto M (2014) Tumor necrosis factor-α enhances RANKL expression in gingival epithelial cells via protein kinase A signaling (in eng). J Periodontal Res 49:508–517. https://doi.org/10.1111/jre.12131
Asano T, Okamoto K, Nakai Y, Tsutsumi M, Muro R, Suematsu A, Hashimoto K, Okamura T, Ehata S, Nitta T, Takayanagi H (2019) Soluble RANKL is physiologically dispensable but accelerates tumour metastasis to bone vol 1. Nat Metabol 1:868–875
Kanzaki H, Makihira S, Suzuki M, Ishii T, Movila A, Hirschfeld J, Mawardi H, Lin X, Han X, Taubman MA, Kawai T (2016) Soluble RANKL cleaved from activated lymphocytes by TNF-α-converting enzyme contributes to osteoclastogenesis in periodontitis (in eng). J Immunol 197:3871–3883. https://doi.org/10.4049/jimmunol.1601114
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA (2011) Matrix-embedded cells control osteoclast formation (in eng). Nat Med 17:1235–1241. https://doi.org/10.1038/nm.2448
Men Y, Wang Y, Yi Y, Jing D, Luo W, Shen B, Stenberg W, Chai Y, Ge WP, Feng JQ, Zhao H (2020) Gli1+ periodontium stem cells are regulated by osteocytes and occlusal force (in eng). Dev Cell 54:639–54.e6. https://doi.org/10.1016/j.devcel.2020.06.006
Takimoto A, Kawatsu M, Yoshimoto Y, Kawamoto T, Seiryu M, Takano-Yamamoto T, Hiraki Y, Shukunami C (2015) Scleraxis and osterix antagonistically regulate tensile force-responsive remodeling of the periodontal ligament and alveolar bone (in eng). Development 142:787–796. https://doi.org/10.1242/dev.116228
Graves DT, Alshabab A, Albiero ML, Mattos M, Corrêa JD, Chen S, Yang Y (2018) Osteocytes play an important role in experimental periodontitis in healthy and diabetic mice through expression of RANKL (in eng). J Clin Periodontol 45:285–292. https://doi.org/10.1111/jcpe.12851
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H (2006) Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction (in eng). J Exp Med 203:2673–2682. https://doi.org/10.1084/jem.20061775
Lin D, Li L, Sun Y, Wang W, Wang X, Ye Y, Chen X, Xu Y (2014) IL-17 regulates the expressions of RANKL and OPG in human periodontal ligament cells via TRAF6/TBK1-JNK/NF-, Äö√¢√†, àö, â†, Äö√Ñ√∂, àö, Ƭ¨¬•B pathways (in eng). Immunology. https://doi.org/10.1111/imm.12395
Yasuhara R, Miyamoto Y, Takami M, Imamura T, Potempa J, Yoshimura K, Kamijo R (2009) Lysine-specific gingipain promotes lipopolysaccharide- and active-vitamin D3-induced osteoclast differentiation by degrading osteoprotegerin (in eng). Biochem J 419:159–166. https://doi.org/10.1042/BJ20081469
Akiyama T, Miyamoto Y, Yoshimura K, Yamada A, Takami M, Suzawa T, Hoshino M, Imamura T, Akiyama C, Yasuhara R, Mishima K, Maruyama T, Kohda C, Tanaka K, Potempa J, Yasuda H, Baba K, Kamijo R (2014) Porphyromonas gingivalis-derived lysine gingipain enhances osteoclast differentiation induced by tumor necrosis factor-α and interleukin-1β but suppresses that by interleukin-17A: importance of proteolytic degradation of osteoprotegerin by lysine gingipain (in eng). J Biol Chem 289:15621–15630. https://doi.org/10.1074/jbc.M113.520510
Ochiai N, Nakachi Y, Yokoo T, Ichihara T, Eriksson T, Yonemoto Y, Kato T, Ogata H, Fujimoto N, Kobayashi Y, Udagawa N, Kaku S, Ueki T, Okazaki Y, Takahashi N, Suda T (2019) Murine osteoclasts secrete serine protease HtrA1 capable of degrading osteoprotegerin in the bone microenvironment (in eng). Commun Biol 2:86. https://doi.org/10.1038/s42003-019-0334-5
Koide M, Kobayashi Y, Ninomiya T, Nakamura M, Yasuda H, Arai Y, Okahashi N, Yoshinari N, Takahashi N, Udagawa N (2013) Osteoprotegerin-deficient male mice as a model for severe alveolar bone loss: comparison with RANKL-overexpressing transgenic male mice (in eng). Endocrinology 154:773–782. https://doi.org/10.1210/en.2012-1928
Tsukasaki M, Asano T, Muro R, Huynh NC, Komatsu N, Okamoto K, Nakano K, Okamura T, Nitta T, Takayanagi H (2020) OPG production matters where it happened (in eng). Cell Rep 32:108124. https://doi.org/10.1016/j.celrep.2020.108124
Cawley KM, Bustamante-Gomez NC, Guha AG, MacLeod RS, Xiong J, Gubrij I, Liu Y, Mulkey R, Palmieri M, Thostenson JD, Goellner JJ, O’Brien CA (2020) Local production of osteoprotegerin by osteoblasts suppresses bone resorption (in eng). Cell Rep 32:108052. https://doi.org/10.1016/j.celrep.2020.108052
Oğütcen-Toller M, Tek M, Sener I, Bereket C, Inal S, Ozden B (2010) Intractable bimaxillary osteomyelitis in osteopetrosis: review of the literature and current therapy (in eng). J Oral Maxillofac Surg 68:167–175. https://doi.org/10.1016/j.joms.2005.07.022
Vivier E, van de Pavert SA, Cooper MD, Belz GT (2016) The evolution of innate lymphoid cells (in eng). Nat Immunol 17:790–794. https://doi.org/10.1038/ni.3459
Reisz RR, Scott DM, Pynn BR, Modesto SP (2011) Osteomyelitis in a Paleozoic reptile: ancient evidence for bacterial infection and its evolutionary significance (in eng). Naturwissenschaften 98:551–555. https://doi.org/10.1007/s00114-011-0792-1
Acknowledgements
I thank H.Takayanagi, N.Komatsu, K.Nagashima, T.Nitta, W.Pluemsakunthai, C.Shukunami, Y.Iwakura, T.Nakashima and K.Okamoto for their great contribution to the publications, on which this work is based. This work was supported in part by a grant for the Young Scientists (19K18943) and JSPS fellows (18J00744) from the Japan Society for the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Tsukasaki, M. RANKL and osteoimmunology in periodontitis. J Bone Miner Metab 39, 82–90 (2021). https://doi.org/10.1007/s00774-020-01165-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00774-020-01165-3