
Abstract
Numerous studies have demonstrated the advantages of plant cell suspension culture systems in producing bioactive recombinant human growth factors. This study investigated the biological activity of recombinant basic human fibroblast growth factor (rhFGF2) protein produced by a plant culture system to enhance new bone formation in a bone defect mouse model. The human FGF2 cDNA gene was cloned into a plant expression vector driven by the rice α-amylase 3D promoter. The vector was introduced into rice calli (Oryza sativa L. cv. Dongjin), and the clone with the highest expression of rhFGF2 was selected. Maximum accumulation of rhFGF2 protein (approximately 28 mg/l) was reached at 13 day post-incubation. Male C57BL/6 mice underwent calvarial defect surgery and the defects were loaded with absorbable collagen sponge (ACS) only (ACS group) or ACS impregnated with 5 μg of plant-derived rhFGF2 (p-FGF2) protein or E. coli-derived rhFGF2 (e-FGF2) protein. Similar to the effects of e-FGF2, local delivery with p-FGF2 enhanced bone healing in the damaged region to higher levels than the ACS group. Exogenous addition of p-FGF2 or e-FGF2 exhibited similar effects on proliferation, mineralization, and osteogenic marker expression in MC3T3-E1 cells. Together, the current findings support the usefulness of this plant-based expression system for the production of biologically active rhFGF2.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bone is a constantly created and replaced tissue that is maintained through a balance between osteoblast and osteoclast activity. However, congenital defects, trauma injury, and tumors can cause large or critical-sized bone defects, which are often beyond the self-regenerative capacity of bone [1, 2]. The implantation of autografts or allografts is currently clinically applied as a standard therapeutic method for large bone defects [3,4,5]. However, bone grafting requires multiple invasive surgeries over long periods of time and is associated with expensive surgical costs [6].
The use of demineralized bone matrix and synthetic biomaterials or in combination with growth factors has been considered an alternative clinical approach for bone regeneration [7,8,9,10,11,12]. Growth factors such as bone morphogenetic proteins (BMPs), epidermal growth factor (EGF), fibroblast growth factors (FGFs), insulin-like growth factors (IGFs), and platelet-derived growth factor have been shown to synergistically stimulate the reconstruction of large bone defects [12,13,14]. Growth factors likely enhance the recovery of injured tissues by directly and/or indirectly regulating multiple cellular events such as survival, differentiation, proliferation, and migration [15, 16]. Among the growth factors, BMPs play important functions in enhancing bone repair and regeneration [17, 18]. FGFs and their receptors are also crucial molecules for the regulation of bone development and homeostasis [19]. Basic FGF (FGF2) is produced and secreted in many types of cells through an endoplasmic reticulum/Golgi-independent secretory route. The activation of FGF2 and its receptor affects multiple biological processes required for wound healing, tumorigenesis, angiogenesis, and tissue remodeling [20,21,22]. Activation of FGF2-mediated signaling also induces osteoblastic differentiation and mineralization, and promotes bone healing by stimulating the mitogen-activated protein kinase-runt-related transcription factor-2 (Runx2) pathway [23,24,25,26]. The previous studies suggested that FGF2 signaling can regulate osteoblastic niche cells to support the homeostasis of hematopoietic stem cells in response to bone marrow damage [12]. These reports strongly indicate that, in addition to BMPs and other FGFs, FGF2 may also be clinically useful in stimulating bone healing and thus enhancing bone regeneration in large or critical-sized bone defects.
As the clinical approaches using growth factors for tissue regeneration and wound healing have gradually expanded, improving the production efficacy and pharmaceutical activity of growth factors that do not show any side effects is critical. Several heterologous expression systems are widely used to produce recombinant growth factors, and DNA recombinant technology using Escherichia coli is the most common system. However, the recent approaches have used a transgenic plant cell suspension culture system for the production of recombinant proteins [27, 28]. Plant cell culture systems are relatively inexpensive and scalable to other expression systems, and the secreted proteins can be efficiently and conveniently purified with reduced contamination risk from viral and bacterial toxins [28,29,30,31]. Indeed, we previously produced IGF-1 using a transgenic rice cell suspension culture system and found that local supplementation of the generated IGF-1 enhanced new bone formation in critical-sized calvarial defects in mice [32]. These results show that plant-based expression systems can provide various advantages over microbial and mammalian cell culture systems in recombinant protein production.
In addition to the improvement of culture systems, many investigators have also attempted to increase the production rate of recombinant proteins and their accumulation in culture medium. The rice amylase 3D (RAmy3D) gene, a member of the rice α-amylase gene family, has long been used for manufacturing recombinant proteins in rice cell suspension culture. The RAmy3D promoter is used to drive expression in plant cell suspension culture and is tightly controlled by sucrose starvation [9, 33]. Numerous studies have demonstrated that the RAmy3D promoter is a powerful production system for recombinant proteins [34,35,36] and shows the advantages derived from plant-based expression system [35].
In this study, we produced recombinant human FGF2 (rhFGF2) protein in transgenic rice cell suspension cultures using the low-cost and high-level RAmy3D α-amylase expression system. We investigated the biological activity of plant-derived rhFGF2 protein, named p-FGF2, to enhance bone healing in a mouse calvarial defect model, as well as its effects on cellular activities including proliferation and osteogenic differentiation of MC3T3-E1 cells. We compared the biological activities of p-FGF2 with that of FGF2 produced using a bacterial (E. coli) expression system (named e-FGF2). To compare the mechanisms by which p-FGF2 and e-FGF2 exert their activities on bone or osteoblasts, the expression patterns of osteoblast-specific markers in newly formed bones of calvarial defects and in MC3T3-E1 pre-osteoblastic cells were examined.
Materials and methods
Chemicals and laboratory equipments
e-FGF2 (≥ 98% purity) was purchased from BioVision (Milpitas, CA, USA). Antibodies specific to osteopontin (OPN; ab8448) and Runx2 (ab23981) were obtained from Abcam (Cambridge, UK), while anti-osteocalcin (OCN; BS7961) was purchased from Bioworld Technology Inc. (St. Louis Park, MN, USA). Fetal bovine serum (FBS) was purchased from HyClone Laboratories (Logan, UT, USA). Unless specified otherwise, chemicals were purchased from Sigma-Aldrich Co. LLC (St. Louis, MO, USA), and laboratory consumables were from Falcon Labware (Becton–Dickinson, Franklin Lakes, NJ, USA).
Construction of expression vector
p-FGF2 was produced using the RAmy3D promoter in the transgenic rice cell suspension culture system as described previously [28, 37]. In brief, a sequence including the human FGF2 gene harboring the rice 3D amylase signal peptide was synthesized using the human FGF2 DNA sequence (NCBI accession number NM 002006) via an overlap PCR strategy (Fig. 1a). The shFGF2 DNA sequence was optimized based on rice codon usage (http://www.kazusa.or.jp/codon). The shFGF2 gene was cloned into the pGEM-T Easy vector (Promega, Madison, WI, USA) and the DNA sequence was confirmed by DNA sequence analysis. The shFGF2 gene was then amplified by PCR and the resultant 613 bp PCR product was introduced into a plant expression vector, pMYN75, containing hygromycin phosphotransferase as a selection marker for plant transformation and the rice RAmy3D promoter expression system according to the methods described previously [38] (Fig. 1b).

Construction of hFGF2-pMYN75. a Overlap PCR strategy for the synthesis of shFGF2. b A schematic diagram of the gene construct used in this study. The human basic fibroblast growth factor (hFGF2) gene harboring the signal peptide of the rice amylase 3D gene is located between the rice amylase 3D promoter and the 3′ untranslated region (3′ UTR). Transferred DNA (T-DNA) of the final plasmid is shown. RB T-DNA right border, 3′ UTR 3′ untranslated region, du35S CaMV35S promoter with a duplicated enhance region, HPT hygromycin phosphotransferase gene, Tnos terminator of nopaline synthase, LB T-DNA left border
Rice transformation and screening of transgenic rice cell lines
Rice calli (Oryza sativa L. cv. Dongin) were prepared and transformed with shFGF2-pMYN75 using a modification of particle bombardment-mediated transformation as described previously [39]. The explants were transferred to N6 selection medium supplemented with 2,4-dichlorophenoxyacetic acid (2 mg/l), sucrose (30 g/l), proline (0.5 g/l), glutamine (0.5 g/l), casein enzymatic hydrolysate (0.3 g/l), gelite (2 g/l), and hygromycin B (35 mg/l) for selection, and then, the explants were transferred onto fresh medium after 2–3 weeks. Callus resistant to hygromycin B was screened for hFGF2 expression by growing in N6 selection medium without sucrose for 3 days. The presence of hFGF2 in the selected callus was analyzed by PCR using gene-specific primers, and rhFGF2 concentration in culture was determined using a human FGF2-specific ELISA kit (Endogene, Woburn, MA, USA) according to the manufacturer’s instruction. Among the 18 transgenic calli, KF40-14 was selected as the optimum cell line for efficient production of rhFGF2 in N6 medium.
Production and purification of p-FGF2
The KF40-14-transformed rice callus was cultured at 25°C in the dark using a shaking incubator with a rotation speed of 110 rpm in 1000 ml of medium supplemented with 2 mg/l of 2,4-dichlorophenoxyacetic acid, 0.02 mg/l of kinetin, and 3% sucrose. Inocula (200 ml) were transferred to new media for culture and the media were changed every 7 days. To induce the expression of hFGF2, N6 medium was removed from the suspension culture by aspiration and the cells were transferred to fresh sucrose-free N6 medium. The supernatant (1000 ml) was collected and the proteins were harvested by filtration. The rhFGF2 protein was purified by heparin-affinity chromatography, quantified by Bradford protein assay (Bio-Rad, Hercules, CA, USA), and identified by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and high-performance liquid chromatography. The purity of rhFGF2 was greater than 95% (data not shown). Purified plant-derived rhFGF2 protein (p-FGF2) was lyophilized (1 mg/vial) and stored at − 70°C before use.
Animals and ethics statement
Male C57BL/6 mice (6 weeks old) were obtained from Orient Bio (Daejeon, South Korea). All mice were equilibrated to the new laboratory environment for 1 week before surgical operation. Animals were housed at 22 ± 1°C and 55 ± 5% humidity on an auto-cycling 12 h light/dark cycle with free access to food and water. This study was carried out in strict accordance with the recommendations in the Guide for the Animal Care and Use of the Chonbuk National University. The study protocol was approved by the Chonbuk National University Committee on Ethics in the Care and Use of Laboratory Animals (CBU 2012-0039). The consumption of food and water and the behavior of the animals were monitored every 12 h per day during the experimental periods.
Establishment of calvarial defect mouse model and treatment groups
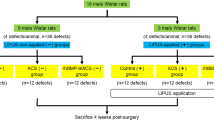
A critical-sized calvarial defect model was established in mice according to previously described methods [32, 40]. Mice were assigned randomly to three experimental groups (n = 18/group). The mean body weights among the groups were similar. Surgical operation was performed on mice (7 weeks old) to create a circular bone defect (4 mm in diameter) at the middle of the sagittal suture. Defects were loaded with an absorbable collagen sponge (ACS; 4 mm diameter and 1 mm thickness) (ACS group) or an ACS impregnated with 15 μl of Dulbecco’s phosphate buffered saline containing 5 µg of p-FGF2 (p-FGF2 group) or e-FGF2 (e-FGF2 group). To analyze the expression patterns of osteogenic marker genes, mice in the three experimental groups (n = 3/group) were sacrificed 2 weeks after surgery and the scaffolds implanted into the calvarial defects were collected.
Real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis
Total RNA was isolated from the scaffolds and real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis was performed. Oligonucleotide primers specific to osteogenic markers, such as Runx2, osterix, OCN, OPN, bone sialoprotein (BSP), and type 1 collagen (Col1A1), were designed to amplify products less than 200 bp using Primer Express 3.0 (Applied Biosystems, Foster City, CA, USA). Primers are listed in Table 1. GAPDH was used as an endogenous reference for quantification.
Immunohistochemistry (IHC) and staining analyses
For IHC staining, calvarial bones including the defect area were removed from the experimental groups (n = 5/group) 2 weeks after surgery. IHC staining was performed using an ImmunoVectastain ABC kit (Vector Laboratories, Inc., Burlingame, CA, USA) following the manufacturer’s instructions. Antibodies specific to Runx2, OCN, and OPN were used at 1:200 dilution, and staining of newly formed bones was observed under a light microscope (Carl Zeiss, Oberkochen, Germany). Mineralization in the calvarial defects of the experimental groups (n = 5/group) was analyzed at 4 weeks after surgery by staining calvarial section samples (5 μm thickness/sample) with Masson’s trichrome. In both IHC and Masson’s trichrome staining, at least five slices per sample were analyzed.
Bone formation analysis
New bone formation in the defect areas of the experimental groups was evaluated by the μCT analysis at 10 week post-surgery (n = 5/group). All procedures for μCT scanning and image analysis followed the methods described elsewhere [32, 40]. Briefly, mice were anesthetized by intramuscular injection with a mixture of zoletil and rompun, and µCT imaging was performed using a SkyScan 1076 microfocus X-ray system (SkyScan®, Kontich, Belgium) with software including NRecon reconstruction®, CTAn 1.8®, and CTvol. The X-ray source was set at 100 kV and 100 µA with a pixel size of 18 µm, a 1-mm filter, and a tomographic rotation of 360° (rotation step of 0.6°). Various bone-specific parameters including bone volume (BV, mm3), bone volume percentage (BV/TV, %), bone surface (BS, mm2), BS in a total tissue volume (BS/TV, 1/mm), structure model index (SMI), trabecular thickness (Tb.th, mm), trabecular separation (Tb.Sp, mm), trabecular number (Tb.N, 1/mm), fractal dimension (FD), total porosity (%), and connectivity density (Conn.Dn, 1/mm3) were calculated in newly formed bone using 3D images reconstructed from the SkyScan NRecon Reconstruction package (Data viewer, Bruker-micro CT-Analyser Ver. 1.13). Bone mineral density (BMD, g/cm3) was calculated by converting the attenuation data for volume of interest into Hounsfield units and BMD units using phantoms (SkyScan) that had a standard density corresponding to mouse bone [32].
Biological effects of rhFGF2 in an in vitro cell culture system
The effects of p-FGF2 and e-FGF2 on the proliferation and osteogenic differentiation of pre-osteoblastic cells were evaluated according to the methods described previously [32, 40]. In brief, MC3T3-E1 cells (ATCC, CRL-2593) were cultured in α-Minimum Essential Medium (α-MEM) supplemented with 10% FBS and antibiotics in 100 mm culture plates and seeded into 96-multiwell culture plates at a density of 2 × 103 cells/well. The culture medium was replaced with serum-free α-MEM 16 h before exposure to various concentrations (0–50 ng/ml) of p-FGF2 or e-FGF2. After incubation for 48 h, 10 μl of Cell Counting Kit-8 (CCK-8) solution (Dojindo Lab., Kumamoto, Japan) was added to each well and absorbance was measured at 450 nm using a microplate reader (Molecular Devices, Austin, TX, USA). In a separate experiment, MC3T3-E1 cells were seeded into 6-well culture plates containing α-MEM supplemented with 10% FBS until the cells reached 90% confluence. The medium was replaced with α-MEM containing p-FGF2 (25 ng/ml), e-FGF2 (25 ng/ml), or DAG (100 nM dexamethasone, 50 μM ascorbic acid, and 5 mM β-glycerophosphate) in the presence of 10% FBS. DAG-treated cells were used as a positive control for mineralization. After 7 days of incubation, the expressions of osteogenic marker genes Runx2, osterix, OCN, OPN, and Col1A1 were determined by real-time RT-PCR analysis. Mineralization of the cells was also measured by staining cells with 0.2% Alizarin red after 21 days of incubation.
Statistical analysis
All experiments were performed at least in triplicate, and data are presented as mean ± standard error of the mean. One-way analysis of variance was used to determine the significance of differences among groups using the Statistical Package for the Social Sciences (SPSS) (version 12.0). When one-way ANOVA was significant (p < 0.05), post hoc Tukey test was used to determine significant differences between groups. Student’s t test was used only when the significance of differences between two sets of data was determined using the SPSS program. A p value < 0.05 was considered statistically significant.
Results
Establishment of FGF2 transgenic rice culture lines
To obtain a rice cell line for high production of p-FGF2 in a transgenic rice cell suspension culture, cDNAs encoding hFGF2 gene with the signal sequence of the rice amylase 3D gene were introduced into a plant expression vector, pMYN75, under the control of the RAmy3D promoter (Fig. 1b). Integration of the hFGF2 gene was confirmed by PCR analysis (data not shown). A total of 18 cell lines were selected for further analyses.
Cell line screening and production and purification of p-FGF2 from transgenic rice suspension culture
The production of p-FGF2 in the 18 selected cell lines was analyzed using an hFGF2 protein-specific ELISA kit to screen for cell lines expressing a high level of p-FGF2. A wide variation in the induced p-FGF2 amount was detected among the transgenic cell lines. The KF40-2 cell line expressed the lowest level of p-FGF2, while the KF40-14 line showed the highest production of p-FGF2 (Supplemental Fig. 1). The KF40-14 cell line was thus selected for further analyses. Next, the maximum production phase of p-FGF2 was determined by performing a time-course experiment (Supplemental Fig. 2). The expression of p-FGF2 was not detected until 7 days into the growth phase. After the medium was changed to a sucrose-free new N6 medium, p-FGF2 concentration in culture medium gradually increased in a time-dependent manner. The maximum production level of p-FGF2 (28 mg/l) was detected at 13 days after culture in sucrose-free N6 culture medium. p-FGF2 was collected 13 days after the incubation and purified before lyophilization. Supplemental Fig. 3 shows the sizes of p-FGF2 and e-FGF2 detected by silver staining and Western blotting.
Local delivery of p-FGF2 increases the expression of osteogenic marker genes in newly formed bone in a calvarial defect mouse model
We next examined the biological effects of p-FGF2 and e-FGF2 to the stimulation of new bone formation using a calvarial bone defect mouse model. Supplemental Fig. 4 shows the procedures for surgery and implantation of ACS alone, ACS impregnated with p-FGF2, or ACS impregnated with e-FGF2 in the calvarial defects. Total RNA was isolated from the ACSs at 2 weeks after surgery and the expression levels of osteogenic marker genes were determined. The p-FGF2 group showed significantly higher levels of Runx2 (p < 0.001), osterix (p < 0.01), BSP (p < 0.001), Col1A1 (p < 0.05), OCN (p < 0.001), and OPN (p < 0.05) compared with the ACS group (Fig. 2). Similarly, the e-FGF2 group revealed higher expression of these genes in the newly formed bone compared to the ACS group. There was no significant difference in the increase in osteogenic marker genes between the plant- and the E. coli-derived FGF2. To examine the effects of p-FGF2 on the induction of osteogenic proteins, we analyzed the protein levels of Runx2, OPN, and OCN in newly formed bone of calvarial defects 2 weeks after surgery (Fig. 3a). Similar to the effects on osteogenic marker gene expression, local delivery of p-FGF2 induced osteogenic marker proteins at the defect site compared with mice that only received ACS. Local supplementation with e-FGF2 also increased the induction of osteogenic proteins to levels similar to those of p-FGF2. The e-FGF2- or p-FGF2-mediated increases in osteogenic protein induction were supported by quantifying the number of cells expressing COX-2, TNF-α, and ICAM-1 after IHC staining (Fig. 3b).

Expression of osteoblast-specific marker genes within the defect margin in the experimental groups. Mice underwent calvarial defect surgery and the defects were loaded with absorbable collagen sponge (ACS) alone (ACS group) or ACS with plant-derived FGF2 (p-FGF2) protein or E. coli-derived FGF2 (e-FGF2). Mice were sacrificed 2 weeks after surgery (n = 3/group) and total RNAs were extracted from the defect sites. Levels of Runx2, osterix, BSP, Col1A1, OCN, and OPN were analyzed by real-time RT-PCR. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the ACS group

Induction of osteoblast-specific marker proteins within the defect margin in the experimental groups. a Two weeks after surgery, the induction of Runx2, OPN, and OCN proteins in the defect areas from the ACS, p-FGF2, and e-FGF2 groups were determined by IHC staining (n = 5/group). Results are representative of five different samples. Bar, 100 μm; Magnification, × 200. b The numbers of IHC positive cells specific to COX-2, TNF-α, or ICAM-1 were counted. **p < 0.01 and ***p < 0.001 compared with the ACS group
Local delivery of p-FGF2 promotes new bone formation in calvarial defects
We examined whether the FGF2-stimulated upregulation in the expression of osteogenic marker genes and proteins was coupled with enhanced bone formation in the region of calvarial defects. To this end, the scaffolds implanted in calvarial defects were isolated 4 weeks after surgery and stained with Masson’s trichrome (Fig. 4). The ACS group did not show new bone formation in any zone of the defect space, whereas both p-FGF2 and e-FGF2 groups showed the induction of mineralized bone in the defect region. The p-FGF2 group tended to show formation of thicker and broader trabecules surrounding the defect regions compared with the e-FGF2 group. We next analyzed the effect of plant- and E. coli-derived FGF2 on bone formation in calvarial defects through μCT analysis at 7 and 10 week post-surgery. The p-FGF2 group showed new bone formation in the region of calvarial defects, even at 7 weeks after surgery, and the p-FGF2-mediated amounts of mineralized bone were greater compared to the ACS group (data not shown). Similarly, both FGF2 groups had greater increases in new bone formation compared with the ACS group at 10 weeks after surgery (Fig. 5a). The FGF2-mediated enhancement in calvarial bone formation was confirmed by 3D model construction of the calvarial defect region (Fig. 5b). The p-FGF2 and e-FGF2 groups showed significantly greater values for bone parameters, including BV, BV/TV, BS, BS/TV, Tb.th, Tb.N, FD, Conn.Dn, and BMD compared to the ACS group after 10 weeks of surgery (Fig. 5c).

Masson trichrome staining of calvarial defect regions in the experimental groups. The maturity of newly formed bone from the ACS, p-FGF2, and e-FGF2 groups was evaluated 4 weeks after surgery. Upper and lower panels show × 25 and × 100 magnified images, respectively. Results are representative of seven different samples. Yellow arrows indicate host bone. S collagen sponge, N new bone within the defect margin

Constructed slice images of calvarial defects in the experimental groups. Mice were subjected to μCT analysis at 10 week post-surgery. Three orthogonal reconstructed slice images (a) and 3D model construction (b) of calvarial defect regions are shown. Red circles indicate the position and dimensions of the 4 mm defect, while the yellow color designates newly ossified tissues within the margin of the defect. Representative results of seven different samples from the ACS control and FGF2 groups are shown. c Various bone-specific parameters, including BV, BV/TV, BS, BS/TV, SMI, Tb.th, Tb.Sp, Tb.N, FD, Po.tot, Conn.Dn, and BMD, were also measured in the ACS, p-FGF2, and e-FGF2 groups. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the ACS group (n = 5)
p-FGF2 treatment inhibits proliferation, but increases bone-specific gene expression and mineralization in MC3T3-E1 cells
To further understand the underlying mechanisms by which local delivery of growth factors enhances bone formation, we explored how exogenous addition of growth factors affected the proliferation and mineralization of bone-like cells. Cell proliferation assay results showed that 50 ng/ml of p-FGF2 or e-FGF2 did not stimulate proliferation, but rather inhibited proliferation of MC3T3-E1 cells (Fig. 6a). Exogenous addition of p-FGF2 or e-FGF2 (25 ng/ml) increased mineralization of the cells, although the number of cells stained with Alizarin red appeared to be lower than that achieved with DAG treatment, the positive control (Fig. 6b). Measuring the optical density corresponding to Alizarin red dye staining also supported greater mineralization in the cells exposed to p-FGF2 or e-FGF2 compared to that in the untreated controls (data not shown). We next examined the expression levels of Runx2, osterix, OCN, OPN, and Col1A1 in MC3T3-E1 cells by real-time RT-PCR analysis after 7 days of incubation with p-FGF2 or e-FGF2. The levels of Runx2, osterix, OCN, and OPN were significantly augmented in the cells treated with DAG (positive control), p-FGF2, or e-FGF2 compared with untreated cells (Fig. 6c). Among these genes, the level of OPN in the cells treated with p-FGF2 or e-FGF2 was significantly higher compared with levels in DAG-treated cells. The expression of Col1A1 in DAG-treated cells was not changed compared to untreated cells, whereas Col1A levels in the cells exposed to p-FGF2 or e-FGF2 were significantly lower, compared to the untreated cells (p < 0.001).

Effects of p-FGF2 and e-FGF2 on proliferation, mineralization, and expression of osteogenic marker genes in MC3T3-E1 cells. a Cells were incubated with 5, 25, and 50 ng/ml of p-FGF2 or e-FGF2, and cell proliferation was evaluated by CCK-8 assay. **p < 0.01 compared with untreated control cells (n = 5). b, c Cells were also incubated in α-MEM containing 10% FBS alone (negative control) or with p-FGF2 (25 ng/ml), e-FGF2 (25 ng/ml), or DAG (positive control). b After 21 days of incubation, mineralization of the cells was measured by staining with 0.2% Alizarin red. A representative result from three different samples is shown. c After 7 days of incubation, cells were examined by real-time RT-PCR analysis. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with untreated control cells;#p < 0.05 compared to DAG-treated cells (n = 5)
Discussion
Accumulating evidence suggests that local delivery of recombinant therapeutic proteins can enhance the processes required for bone and tissue healing [17, 18, 41]. FGF2 promotes the processes required for bone healing by controlling multiple cellular events such as mobilization, proliferation, and differentiation of osteoblast lineages cells [31], as well as by upregulating the expression of osteoblast-specific marker genes [23, 24]. Numerous studies have demonstrated that local administration of FGF2 stimulates osteogenic gene expression and bone formation in damaged bones [42,43,44]. These reports strongly support the potentials of FGF2 in clinical use for bone reconstruction.
Various heterologous expression systems have been developed to produce recombinant proteins, and bacteria such as E. coli or Bacillus subtilis are the most common expression systems [45, 46]. E. coli provide several benefits for the production of recombinant proteins such as low cost and low risk of contamination with endotoxin. However, in some cases, E. coli systems fail to produce complex proteins due to protein incorporation into inclusion bodies, the development of disulfide-linked aggregates, and the requirements of specific post-translational modifications of the produced proteins. The E. coli system is also not completely free from contamination with toxins, even if the protein of interest is being purified by SDS-PAGE and/or chromatography. Mammalian cells are another useful system for complex protein production with post-translational modification, but this system can be associated with increased production costs. Furthermore, yeast systems show limitations due to the difference in glycosylation patterns [47]. Transgenic plant cell suspension culture system using soybean or rice is a relatively efficient and convenient expression system that can overcome the disadvantages encountered with E. coli, mammalian cells, or yeast [48,49,50,51,52]. The plant cell-based expression system is also free from animal pathogens and viruses, and can allow for large mass production of recombinant proteins in a sterilized- and environmental-controlled bioreactor at a relatively low cost compared to E. coli [52,53,54,55]. In addition, the plant-based expression system uses natural protein storage organs and post-translational modification patterns similar to that of humans, and recombinant proteins can be isolated from culture supernatants without a cell lysis step [52,53,54,55]. Furthermore, recombinant human proteins produced by the plant cell culture system can be preserved in a mature form with more biologically active conformation compared to protein produced by E. coli systems [32]. Of note, FGF2 is a non-glycosylated protein, and thus, the activity and bioavailability of FGF2 is independent of post-translational modifications.
Minimizing production costs is crucial for the establishment of transgenic plant cell culture systems. According to a previous report [56], the production cost to yield a recombinant protein using the transgenic plant cell culture system is up to 50-fold lower than producing the same amount of recombinant protein in a mammalian cell culture system. Consequently, this study together with our previous report [32] strongly supports the usefulness and efficiency of using transgenic rice cell suspension culture systems as bioreactors to produce recombinant human growth factors as well as the other therapeutic proteins. Furthermore, our study findings demonstrate that the bioactivity of the plant-derived FGF2 is similar to that of E. coli-derived FGF2. This indicates that the plant-derived growth factors including FGF2 may be clinically useful in treating large- and critical-sized bone defects.
Runx2 and osterix tightly regulate osteoblast differentiation and mineralization. These transcription factors bind to the promoter regions of osteoblast-specific genes, and control the expression of downstream osteogenic marker genes such as ALP, BSP, OCN, OPN, Col1A1, and osteonectin genes [57,58,59]. Our results showed that local treatment with p-FGF2 using ACS enhanced Runx2 and osterix mRNA and protein levels with concomitant increases in downstream effectors such as BSP, Col1A1, OCN, and OPN in the region of calvarial defects, as well as in cultured pre-osteoblast cells. Collectively, it is likely that determining the expression levels of osteoblast-specific markers is the most common approach to understand the molecular mechanisms by which an interested biomaterial positively affects bone formation.
In summary, to the best of our knowledge, we describe here the convenient and efficient production of rhFGF2 in a transgenic rice cell suspension culture with the rice α-amylase RAmy3D expression system. Our results indicate the possibility that plant-derived recombinant proteins may be useful in clinical approaches for bone regeneration, wound healing, and cosmetic treatment. The previous studies have demonstrated the synergistic effects of FGF2 in combination with BMPs or other bone-specific biomaterials for inducing greater therapeutic effects in clinical than FGF2 treatment alone [15, 50]. Further studies to verify the biological effects of p-FGF2 in the combined treatment with another biomaterial will be needed.
References
Aghaloo T, Cowan CM, Zhang X, Freymiller E, Soo C, Wu B, Ting K, Zhang Z (2010) The effect of NELL1 and bone morphogenetic protein-2 on calvarial bone regeneration. J Oral Maxillofac Surg 68:300–308
Huang RL, Kobayashi E, Liu K, Li Q (2016) Bone graft prefabrication following the in vivo bioreactor principle. EBioMedicine 12:43–54
Bhumiratana S, Vunjak-Novakovic G (2012) Concise review: personalized human bone grafts for reconstructing head and face. Stem Cells Transl Med 1:64–69
Oryan A, Alidadi S, Moshiri A (2013) Current concerns regarding healing of bone defects. Hard Tissue 2:13
Papageorgiou SN, Papageorgiou PN, Deschner J, Götz W (2016) Comparative effectiveness of natural and synthetic bone grafts in oral and maxillofacial surgery prior to insertion of dental implants: systematic review and network meta-analysis of parallel and cluster randomized controlled trials. J Dent 48:1–8
Amini AR, Laurencin CT, Nukavarapu SP (2012) Bone tissue engineering: recent advances and challenges. Crit Rev Biomed Eng 40:363–408
Calori GM, Mazza E, Colombo M, Ripamonti C (2011) The use of bone-graft substitutes in large bone defects: any specific needs? Injury 42:S56–S63
Dimitriou R, Jones E, McGonagle D, Giannoudis PV (2011) Bone regeneration: current concepts and future directions. BMC Med 9:66
Simmons CR, Huang N, Cao Y, Rodriguez RL (1991) Synthesis and secretion of α-amylase by rice callus: evidence for differential gene expression. Biotechnol Bioeng 38:545–551
Vaccaro AR (2002) The role of the osteoconductive scaffold in synthetic bone graft. Orthopedics 25:S571–S578
Vo TN, Kasper FK, Mikos AJ (2012) Strategies for controlled delivery of growth factors and cells for bone regeneration. Adv Drug Deliv Rev 64:1292–1309
Yoon KA, Son Y, Choi YJ, Kim JH, Cho JY (2017) Fibroblast growth factor 2 supports osteoblastic niche cells during hematopoietic homeostasis recovery after bone marrow suppression. Cell Commun Signal 15:25
Chen X, Wang J, Yu L, Zhou J, Zheng D, Zhang B (2018) Effect of concentrated growth factor (CGF) on the promotion of osteogenesis in bone marrow stromal cells (BMSC) in vivo. Sci Rep 8:5876
Simpson AH, Mills L, Noble B (2006) The role of growth factors and related agents in accelerating fracture healing. J Bone Jt Surg 88:701–705
Gothard D, Smith EL, Kanczler JM, Rashidi H, Qutachi O, Henstock J, Rotherham M, El Haj A, Shakesheff KM, Oreffo RO (2014) Tissue engineered bone using select growth factors: a comprehensive review of animal studies and clinical translation studies in man. Eur Cells Mater 28:166–207
Lieberman JR, Daluiski A, Einhorn TA (2002) The role of growth factors in the repair of bone. Biology and clinical applications. J Bone Jt Surg Am 84-A:1032–1044
Anusuya GS, Kandasamy M, Jacob Raja SA, Sabarinathan S, Ravishankar P, Kandhasamy B (2016) Bone morphogenetic proteins: signaling periodontal bone regeneration and repair. J Pharm Bioallied Sci 8:S39–S41
Salazar VS, Gamer LW, Rosen V (2016) BMP signalling in skeletal development, disease and repair. Nat Rev Endocrinol 12:203–221
Charoenlarp P, Rajendran AK, Iseki S (2017) Role of fibroblast growth factors in bone regeneration. Inflamm Regen 37:10
Bikfalvi A, Klein S, Pintucci G, Rifkin DB (1997) Biological roles of fibroblast growth factor-2. Endocr Rev 18:26–45
Su N, Jin M, Chen L (2014) Role of FGF/FGFR signaling in skeletal development and homeostasis: learning from mouse models. Bone Res 2:14003
Szebenyi G, Fallon JF (1999) Fibroblast growth factors as multifunctional signaling factors. Int Rev Cytol 185:45–106
Fei Y, Hurley MM (2012) Role of fibroblast growth factor 2 and Wnt signaling in anabolic effects of parathyroid hormone on bone formation. J Cell Physiol 227:3539–3545
Montero A, Okada Y, Tomita M, Ito M, Tsurukami H, Nakamura T, Doetschman T, Coffin JD, Hurley MM (2000) Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J Clin Investig 105:1085–1093
Sobue T, Naganawa T, Xiao L, Okada Y, Tanaka Y, Ito M, Okimoto N, Nakamura T, Coffin JD, Hurley MM (2005) Over-expression of fibroblast growth factor-2 causes defective bone mineralization and osteopenia in transgenic mice. J Cell Biochem 95:83–94
Franceschi RT, Xiao G, Jiang D, Gopalakrishnan R, Yang S, Reith E (2003) Multiple signaling pathways converge on the Cbfa1/Runx2 transcription factor to regulate osteoblast differentiation. Connect Tissue Res 44(Suppl 1):109–116
Fischer R, Emans N (2000) Molecular farming of pharmaceutical proteins. Transgenic Res 9:279–299
Shin YJ, Hong SY, Kwon TH, Yang MS (2003) High Level of expression of recombinant human granulocyte-macrophage colony stimulating factor in transgenic rice cell suspension culture. Biotechnol Bioeng 82:778–783
Goldstein DA, Thomas JA (2004) Biopharmaceuticals derived from genetically modified plants. QJM 97:705–716
Jin T, Wang J, Zhu X, Xu Y, Zhou X, Yang L (2015) A new transient expression system for large-scale production of recombinant proteins in plants based on air-brushing an Agrobacterium suspension. Biotechnol Rep 6:36–40
Jung JW, Kim NS, Jang SH, Shin YJ, Yang MS (2016) Production and characterization of recombinant human acid α-glucosidase in transgenic rice cell suspension culture. J Biotechnol 226:44–53
Poudel SB, Bhattarai G, Kook SH, Shin YJ, Kwon TH, Lee SY, Lee JC (2017) Recombinant human IGF-1 produced by transgenic plant cell suspension culture enhances new bone formation in calvarial defects. Growth Horm IGF Res 36:1–10
Huang N, Chandler J, Thomas B, Koizumi N, Rodriguez R (1993) Metabolic regulation of α-amylase gene expression in transgenic cell cultures of rice. Plant Mol Biol 23:737–747
Chung ND, Kim NS, Giap DV, Jang SH, Oh SM, Jang SH, Kim TG, Jang YS, Yang MS (2014) Production of functional human vascular endothelial growth factor 165 in transgenic rice cell suspension cultures. Enzyme Microb Technol 63:58–63
Kim NS, Yu HY, Chung ND, Shin YJ, Kwon TH, Yang MS (2011) Production of functional recombinant bovine trypsin in transgenic rice cell suspension cultures. Protein Expr Purif 76:121–126
Kim BG, Kim SH, Kim NS, Huy NX, Choi YS, Lee JY, Jang YS, Yang MS, Kim TG (2014) Production of monoclonal antibody against FimA protein from Porphyromonas gingivalis in rice cell suspension culture. Plant Cell Tiss Organ Cult 118:293–304
Kim TG, Back MY, Lee EK, Yang MS (2008) Expression of human growth hormone in transgenic rice cell suspension culture. Plant Cell Rep 27:885–891
Hoekema A, Hirsch PR, Hooykaas PJJ, Schieroort RA (1983) A binary plant vector strategy based on separation of vir- and T-region of Agrobacterium tumefaciens Ti-plasmid. Nature 303:179–180
Chen L, Marmey P, Taylor NJ, Brizard JP, Espinoza C, D’Cruz P, Huet H, Zhang S, de Kochko A, Beachy RN, Fauquet CM (1998) Expression and inheritance of multiple transgenes in rice plants. Nat Biotechnol 16:1060–1064
Lim SS, Kook SH, Bhattarai G, Cho ES, Seo YK, Lee JC (2015) Local delivery of COMP-angiopoietin 1 accelerates new bone formation in rat calvarial defects. J Biomed Mater Res A 103:2942–2951
Fowlkes JL, Thrailkill KM, Liu L, Wahl EC, Bunn RC, Cockrell GE, Perrien DS, Aronson J, Lumpkin CK Jr (2006) Effects of systemic and local administration of recombinant human IGF-I (rhIGF-I) on de novo bone formation in an aged mouse model. J Bone Miner Res 21:1359–1366
Furuya H, Tabata Y, Kaneko K (2014) Bone regeneration for murine femur fracture by gelatin hydrogels incorporating basic fibroblast growth factor with different release profiles. Tissue Eng Part A 20:1531–1541
Kodama N, Nagata M, Tabata Y, Ozeki M, Ninomiya T, Takagi R (2009) A local bone anabolic effect of rhFGF2-impregnated gelatin hydrogel by promoting cell proliferation and coordinating osteoblastic differentiation. Bone 44:699–707
Murakami H, Nakasa T, Ishikawa M, Adachi N, Ochi M (2016) Autologous bone grafts with MSCs or FGF-2 accelerate bone union in large bone defects. J Orthop Surg Res 11:105
An N, Ou J, Jiang D, Zhang L, Liu J, Fu K, Dai Y, Yang D (2013) Expression of a functional recombinant human basic fibroblast growth factor from transgenic rice seeds. Int J Mol Sci 14:3556–3567
Wang YP, Wei ZY, Zhong XF, Lin CJ, Cai YH, Ma J, Zhang YY, Liu YZ, Xing SC (2016) Stable expression of basic fibroblast growth factor in chloroplasts of tobacco. Int J Mol Sci 17:19
Upadhyay AK, Murmu A, Singh A, Panda AK (2012) Kinetics of inclusion body formation and its correlation with the characteristics of protein aggregates in Escherichia coli. PLoS One 7:e33951
Aviezer D, Brill-almon E, Shaaltiel Y, Hashmueli S, Bartfeld D, Mizrachi S, Liberman Y, Freeman A, Zimran A, Galun E (2009) A plant-derived recombinant human glucocerebrosidase enzyme—a preclinical and phase I investigation. PLoS One 4:e4792
Beutler E (2006) Lysosomal storage disease: natural history and ethical and economic aspects. Mol Genet Metab 88:208–215
Hughes-Fulford M, Li CF (2011) The role of FGF-2 and BMP-2 in regulation of gene induction, cell proliferation and mineralization. J Orthop Surg Res 6:8
He X, Galpin JD, Tropak MB, Mahuran D, Haselhorst T, Von Itzstein M, Kolarich D, Packer NH, Miao Y, Jiang L, Grabowski GA, Clarke LA, Kermode AR (2012) Production of active human glucocerebrosidase in seeds of Arabidopsis thaliana complex-glycan-deficient (cgl) plants. Glycobiology 22:492–503
Hellwig S, Drossard J, Twyman RM, Fischer R (2004) Plant cell cultures for the production of recombinant proteins. Nat Biotech 22:1415–1422
Giddings G, Allison G, Brooks D, Carter A (2000) Transgenic plants as factories for biopharmaceuticals. Nat Biotechnol 18:1151–1155
Kuo YC, Tan CC, Ku JT, Hsu WC, Su SC, Lu CA, Huang LF (2013) Improving pharmaceutical protein production in Oryza Sativa. Int J Mol Sci 14:8719–8739
Santos RB, Abranches R, Fischer R, Sack M, Holland T (2016) Putting the spotlight back on plant suspension cultures. Front Plant Sci 7:297
Doran PM (2006) Foreign protein degradation and instability in plants and plant tissue cultures. Trends Biotechnol 24:426–432
Choi JY, Lee BH, Song KB (1996) Expression patterns of bone-related proteins during osteoblastic differentiation in MC3T3-E1 cells. J Cell Biochem 61:609–618
Lian JB, Stein GS (2003) Runx2/Cbfa1: a multifunctional regulator of bone formation. Curr Pharm Des 9:2677–2685
Park SS, Kim KA, Lee SY, Lim SS, Jeon YM, Lee JC (2012) X-ray radiation at low doses stimulates differentiation and mineralization of mouse calvarial osteoblasts. BMB Rep 45:571–576
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2016R1A2A2A05921669) and by the Ministry of Education (2018R1D1A1B07046563).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors state that they have no conflict of interests.
Ethical standards
This study was carried out in strict accordance with the recommendations in the Guide for the Animal Care and Use of the Chonbuk National University. The study protocol was approved by the Chonbuk National University Committee on Ethics in the Care and Use of Laboratory Animals (CBU 2012-0039). The consumption of food and water and the behavior of the animals were monitored every 12 h per day during the experimental periods.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig.
1. Screening of transformed callus lines expressing high levels of p-FGF2. The amount of p-FGF2 was determined by ELISA as described in the Materials and Methods. (TIFF 644 kb)
Supplementary Fig.
2. Time-course study of rhFGF2 production in transgenic rice suspension culture medium by direct ELISA. The selected rice cell line (KF40-14) was propagated in sucrose-contained N6 medium and after 9 days of incubation, the medium was replaced to sucrose-free new N6 medium followed by 19 day additional incubation. At the indicated days of incubation, the level of rhFGF2 proteins was monitored by ELISA. (TIFF 890 kb)
Supplementary Fig.
3. Silver staining and Western blot data of p-EGF2 and e-FGF2. The size of p-FGF2 was determined according to silver staining (A) and Western blotting methods (B) by loading 1 μg of the protein (purity > 98%), where e-FGF (1 μg, Prospec CYT-218) was used as positive control. Sliver staining was performed under 14% Tricine reducing condition, while, in immunoblotting, anti-hFGF2 antibody (MyBioSource.com, MBS175351, 2 μg/10 ml) and anti-rabbit IgG antibody were used as 1st and 2nd antibodies, respectively. M, molecular size marker; P, positive control; R, p-FGF2 (original size: 17.2 kDa). (TIFF 2180 kb)
Supplementary Fig.
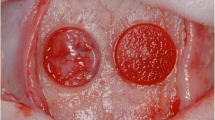
4. The procedures for surgery and ACS implantation. (A) Mice received surgical operation to create calvarial bone defect before implantation with ACS loaded with DPBS, p-FGF2, or e-FGF2. Panels B and C show circular bone defect (4 mm in diameter) at the middle of the sagittal suture and ACS, respectively. (TIFF 3845 kb)
About this article

Cite this article
Poudel, S.B., Min, CK., Lee, JH. et al. Local supplementation with plant-derived recombinant human FGF2 protein enhances bone formation in critical-sized calvarial defects. J Bone Miner Metab 37, 900–912 (2019). https://doi.org/10.1007/s00774-019-00993-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00774-019-00993-2