
Abstract
Autosomal recessive hypophosphatemic rickets (ARHR) is an extremely rare disorder of autosomal recessive inheritance, characterized by hypophosphatemia resulting from renal phosphate wasting. Dentin matrix protein 1 (DMP1), a noncollagenous extracellular protein, plays critical roles in bone mineralization and phosphate homeostasis. Recently, loss-of-function mutations in DMP1 gene have been identified as the molecular cause of ARHR. Here, we describe a Japanese family that includes two ARHR-affected siblings carrying a novel mutation of the DMP1 gene. The patients were a 53-year-old woman and a 50-year-old man with short stature and skeletal deformities who were the offspring of a first-cousin marriage. Biochemical examination revealed hypophosphatemia with renal phosphate excretion and low levels of 1,25(OH)2D. Serum calcium, parathyroid hormone, and urinary calcium excretion were within the normal range, leading to clinical diagnosis of ARHR. Sequence analysis of peripheral leukocytes from the patients revealed that they carried a novel homozygous nonsense mutation in the DMP1 gene (98G>A, W33X), which leads to a truncated DMP protein with no putative biological function. Unaffected family members were heterozygous for the mutation. This is the first report of a Japanese family with ARHR carrying a novel mutation of the DMP1 gene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypophosphatemic rickets is a disorder associated with impaired bone mineralization caused by phosphate deficiency, resulting in skeletal deformity and growth retardation in childhood. Inherited hypophosphatemic rickets is caused by genetic defects that lead to renal phosphate wasting. To date, four types of hypophosphatemic rickets have been identified: X-linked hypophosphatemia (XLH), autosomal dominant hypophosphatemic rickets (ADHR), hereditary hypophosphatemic rickets with hypercalciuria (HHRH), and autosomal recessive hypophosphatemic rickets (ARHR) [1]. Fibroblast growth factor 23 (FGF23), a phosphaturic hormone, has been found to be elevated in XLH [2], ADHR [3, 4], and ARHR [5–7] and is involved in hypophosphatemia in these disorders.
ARHR (OMIM 241520) is an extremely rare disorder of autosomal recessive inheritance, with only a few families of Middle Eastern and European origins reported. ARHR patients exhibit growth retardation, lower-extremity deformities, and dental defects. Later in life, some patients also develop enthesopathy. Patients also show hypophosphatemia as a consequence of renal phosphate wasting and low levels of serum 1,25-dihydroxyvitamin D [1,25(OH)2D], whereas serum calcium, parathyroid hormone (PTH), and urinary calcium excretion are normal. Notably, circulating levels of FGF23 are elevated in patients with ARHR [5–7]. ARHR shares clinical and biochemical features with XLH and ADHR, but recent genetic studies have shown that mutations in the PHEX [8], FGF23 [9], and DMP1 [5, 6] genes underlie XLH, ADHR, and ARHR, respectively, enabling us to distinguish ARHR from other disorders.
Dentin matrix protein 1 (DMP1) is a noncollagenous extracellular matrix protein that is a member of the short integrin-binding ligand-interacting glycoprotein (SIBLING) family [10]. DMP1 is highly expressed in bone and teeth [11] and promotes mineralization [12] and odontogenic differentiation [13]. It has recently been reported that mutations in the DMP1 gene, which is important for phosphate homeostasis, cause ARHR [5, 6].
Here, we examined the DMP1 gene in a Japanese family with ARHR and identified a novel nonsense mutation. This is the first report describing a Japanese family with ARHR carrying a novel mutation of the DMP1 gene.
Patients and methods
Patients
Patient 1 (II-3, Fig. 1) was a 53-year-old woman who had genu varum and growth retardation in childhood. She received no medications, including phosphorus or vitamin D supplementation. At the age of 44 years, she was referred to a hospital because of cervical myelopathy caused by ossification of the posterior longitudinal ligament (OPLL) and was diagnosed with hypophosphatemic rickets. She was 130 cm tall, and had hypophosphatemia (2.2 mg/dl; normal range, 2.7–4.5) with renal phosphate excretion [tubular maximum rate for phosphate reabsorption per glomerular filtrate (TmP/GFR), 1.9 mg/dl; normal range, 2.3–4.5] and low 1,25(OH)2D level (16 pg/ml; normal range, 20–60) (Table 1). Serum calcium, intact PTH, and urinary calcium excretion were within the normal range. She underwent laminoplasty for OPLL and was treated with alfacalcidol for hypophosphatemia. At the age of 53 years, she was admitted to our hospital for hypercalcemia (15.6 mg/dl) and renal insufficiency (serum creatinine, 3.34 mg/dl). She had mild bowing of her legs and severe kyphosis and had lost almost all her teeth (Fig. 2a–c). The high level of serum calcium returned to the normal range, but renal dysfunction was still present (creatinine clearance, 24.4 ml/min) (see Table 1). The serum level of intact FGF23 in patient 1 was extremely high (4540.7 pg/ml; reference range, 10.0–50.0), while the serum creatinine level had declined to 1.15 mg/dl (Table 1).

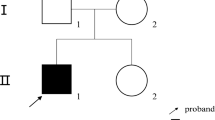
Pedigree of the Japanese family with autosomal recessive hypophosphatemic rickets (ARHR). Black symbols indicate affected individuals. An arrow indicates the proband (patient 1). Half symbols indicate heterozygous individuals. Double line shows a consanguineous marriage. Final heights are shown below the individual symbols. NE, not examined for mutations

Skeletal and dental findings in patient 1 at the age of 53 years. a, b Radiographs show mild bowing of her legs and severe kyphosis. c A panoramic radiograph shows loss of all her teeth
Patient 2 (II-4), a 50-year-old man, was the brother of patient 1. He had genu varum and growth retardation in childhood but received no medications. At the age of 40 years, he was referred to a hospital because of cervical myelopathy caused by OPLL and was diagnosed with hypophosphatemic rickets. He was 145 cm tall, and had hypophosphatemia (2.3 mg/dl) with renal phosphate excretion (TmP/GFR, 2.2 mg/dl) and normal level of 1,25(OH)2D (22 pg/ml). Serum calcium, intact PTH levels, and urinary calcium excretion were within the normal range (see Table 1). He underwent laminoplasty for OPLL and was treated with alfacalcidol for hypophosphatemia. At the age of 50 years, his serum intact FGF23 level was within the normal range (31.8 pg/ml).
The parents of patients I-1 and I-2 were first cousins. The siblings of the patients did not show any clinical evidence of rickets or osteomalacia. Laboratory data of an unaffected sibling (II-2) and a child (III-1) confirmed normophosphatemia (see Table 1). Based on the clinical features, autosomal recessive inheritance, and the absence of hypercalciuria, the patients were diagnosed with ARHR.
Urinary calcium and phosphorus levels were measured after the patients included approximately 500 mg/day calcium and 1000 mg/day phosphorus in their diets for 1 week. Tubular maximum rate for phosphate reabsorption per glomerular filtrate (TmP/GFR) was calculated using a nomogram [14]. Serum intact FGF23 levels were measured as described previously [15].
Mutation analysis
Genomic DNA was extracted from blood samples using the QIAamp DNA Mini Kit (QIAGEN, Tokyo, Japan), according to the manufacturer’s protocol. The five coding DMP1 exons 2–6 were amplified by polymerase chain reaction (PCR) with intronic primers designed with Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi), as described previously [16] (Table 2). PCR products were electrophoresed on an agarose gel, purified from the gel using the UltraClean 15 DNA Purification Kit (MO BIO Laboratories, Solana Beach, CA, USA), and directly sequenced by a BigDye Terminator v3.1 Cycle Sequencing Kit and the ABI PRISM 3100 Genetic Analyzer (Applied Biosystems Japan, Tokyo, Japan). The study was approved by the Ethical Committee of Miyazaki Medical College, and written informed consent was obtained from all subjects before participation.
Results
We performed sequence analysis of DMP1, the causative gene of ARHR. We found that patients 1 and 2 carried a novel homozygous mutation, an A-to-G substitution at nucleotide 98 in exon 3 (98G>A; Fig. 3a). This substitution introduces a premature termination codon (PTC) at codon 33 that replaces the wild-type tryptophan residue (W33X; Fig. 3a). An unaffected sibling (II-2) and the child of patient 1 (III-1) were heterozygous for the mutation.

Sequence analysis of the DMP1 gene. a DNA sequencing reveals a homozygous A-to-G substitution at nucleotide 98 (98G>A, arrows) in ARHR patients (right panel). This substitution introduces a premature termination codon at codon 33 (boxed) that replaces the wild-type tryptophan residue (W33X). b Schematic structure of the DMP1 gene. Mutations that were previously reported and identified in our study are shown
Discussion
DMP1 was originally identified as an extracellular matrix protein [17] and belongs to the SIBLING family, which includes sialoprotein (BSP), osteopontin (OPN), matrix extracellular phosphoglycoprotein (MEPE), and dentin sialophosphoprotein (DSPP) [10]. DMP1 contains a large number of acidic domains that play an important role in mineralization by nucleating the formation of hydroxyapatite crystals [12]. In addition, DMP1 plays a regulatory role in odontogenic differentiation [13]. Recent studies showed that DMP1 null mice exhibit hypophosphatemic rickets, indicating that DMP1 is important for phosphate homeostasis in vivo [5]. In humans, loss-of-function mutations in DMP1 gene were identified as the molecular cause of ARHR, but only a few pathogenic mutations have been reported (Fig. 3b) [5–7].
Here, we identified a novel nonsense mutation (98G>A, W33X) in the DMP1 gene in a Japanese family. It is plausible that the W33X mutation leads to loss of function, because the majority of mRNAs containing premature termination codons (PTCs) are degraded by nonsense-mediated mRNA decay (NMD) to prevent the production of deleterious truncated proteins [18]. As a general rule, PTCs are located more than 50–55 nucleotides upstream of the most 3′ exon–exon junction of the mRNA trigger NMD. In our case, the PTC introduced by the 98G>A mutation is located 84 nucleotides upstream of the most 3′ exon–exon junction, which is between nucleotide 183 (exon 5) and 184 (exon 6). Therefore, the mutant mRNA is likely degraded by NMD. Even if the mutant mRNA is translated, the truncated protein containing only 32 of the 513 amino acids in the full-length protein is assumed to be nonfunctional because it lacks integrin/DNA-binding domains and essential domains for hydroxyapatite nucleation [12, 17, 19].
FGF23 is a bone-derived hormone that stimulates renal phosphate excretion, inhibits the production of 1,25(OH)D2 [20], and is involved in phosphate disorders. Tumor-induced osteomalacia, a paraneoplastic syndrome causing hypophosphatemia, is caused by excess circulating FGF23 derived from mesenchymal tumors [2, 21]. On the other hand, the inactivation of FGF23 results in hyperphosphatemia, such as tumoral calcinosis [22, 23]. Serum phosphate is a major regulator of circulating FGF23, and serum FGF23 levels are markedly elevated in patients with chronic kidney disease [24]. In patient 1, high levels of serum FGF23 levels might be caused by renal insufficiency. However, the serum level of phosphate was inappropriately low in spite of renal sufficiency and treated with oral administration of vitamin D. According to the previous report [25], circulating FGF23 was suppressed to undetectable levels in patients with FGF23-independent hypophosphatemia, such as vitamin D deficiency and Fanconi’s syndrome. Hypophosphatemia among patients with XLH, ADHR, is also suggested to be caused by excess plasma levels of FGF23 [20]. The relatively high circulating FGF23 level in patient 2 and extremely high circulating FGF23 level in patient 1 with hypophosphatemic ARHR suggested the possibility that the high level of circulating FGF23 was a primary origin of hypophosphatemia in patients 1 and 2 with ARHR. A normal level of circulating FGF23 was reported in the patients with ARHR [5, 6], suggesting that circulating FGF23 is not always suppressed in patients with ARHR.
According to previous reports, ARHR is likely to have a wide spectrum of severity. The patients with ARHR in this family were not diagnosed with hypophosphatemia until adult life, probably because their symptoms were mild. On the other hand, other patients underwent repeated osteotomy [6] and developed complications associated with nerve deafness and learning disability. However, a large deletion encompassing at least 49 kb was found in these patients, suggesting the phenotype is not simply caused by the loss of DMP1 [7]. Despite receiving no medications, including vitamin D or phosphorus supplementation, they showed mild skeletal and dental symptoms deformities during adolescence. In addition, patient 1 was able to give birth to three healthy children. The ARHR patients reported here had a mild clinical course and normal fertility without any treatment.
In conclusion, this is the first report of a Japanese family with ARHR. We identified a novel nonsense mutation in the DMP1 gene as the likely cause of the disease. Further accumulation of ARHR cases is necessary to analyze genotype–phenotype correlation.
References
Bastepe M, Jüppner H (2008) Inherited hypophosphatemic disorders in children and the evolving mechanisms of phosphate regulation. Rev Endocr Metab Disord 9:171–180
Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Jüppner H (2003) Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 348:1656–1663
White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ (2001) Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int 60:2079–2086
Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2002) Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology 143:3179–3182
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Lorenz-Depiereux B, Bastepe M, Benet-Pagès A, Amyere M, Wagenstaller J, Müller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Jüppner H, Strom TM (2006) DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet 38:1248–1250
Farrow EG, Davis SI, Ward LM, Summers LJ, Bubbear JS, Keen R, Stamp TC, Baker LR, Bonewald LF, White KE (2009) Molecular analysis of DMP1 mutants causing autosomal recessive hypophosphatemic rickets. Bone (NY) 44:287–294
The HYP Consortium (1995) A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet 11:130–136
ADHR Consortium (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26:345–348
Fisher LW, Torchia DA, Fohr B, Young MF, Fedarko NS (2001) Flexible structures of SIBLING proteins, bone sialoprotein, and osteopontin. Biochem Biophys Res Commun 280:460–465
Qin C, D’Souza R, Feng JQ (2007) Dentin matrix protein 1 (DMP1): new and important roles for biomineralization and phosphate homeostasis. J Dent Res 86:1134–1141
He G, Dahl T, Veis A, George A (2003) Nucleation of apatite crystals in vitro by self-assembled dentin matrix protein 1. Nat Mater 2:552–558
Narayanan K, Srinivas R, Ramachandran A, Hao J, Quinn B, George A (2001) Differentiation of embryonic mesenchymal cells to odontoblast-like cells by overexpression of dentin matrix protein 1. Proc Natl Acad Sci USA 98:4516–4521
Walton RJ, Bijvoet OL (1975) Nomogram for derivation of renal threshold phosphate concentration. Lancet 2:309–310
Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S (2002) Increased circulatory level of biologically active full-length FGF-23 in hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab 87:4957–4960
Beattie ML, Kim JW, Gong SG, Murdoch-Kinch CA, Simmer JP, Hu JC (2006) Phenotypic variation in dentinogenesis imperfecta/dentin dysplasia linked to 4q21. J Dent Res 85:329–333
George A, Sabsay B, Simonian PA, Veis A (1993) Characterization of a novel dentin matrix acidic phosphoprotein. Implications for induction of biomineralization. J Biol Chem 268:12624–12630
Maquat LE (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5:89–99
Narayanan K, Gajjeraman S, Ramachandran A, Hao J, George A (2006) Dentin matrix protein 1 regulates dentin sialophosphoprotein gene transcription during early odontoblast differentiation. J Biol Chem 281:19064–19071
Liu S, Quarles LD (2007) How fibroblast growth factor 23 works. J Am Soc Nephrol 18:1637–1647
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2001) Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA 98:6500–6505
Larsson T, Yu X, Davis SI, Draman MS, Mooney SD, Cullen MJ, White KE (2005) A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab 90:2424–2427
Araya K, Fukumoto S, Backenroth R, Takeuchi Y, Nakayama K, Ito N, Yoshii N, Yamazaki Y, Yamashita T, Silver J, Igarashi T, Fujita T (2005) A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab 90:5523–5527
Larsson T, Nisbeth U, Ljunggren O, Jüppner H, Jonsson KB (2003) Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 64:2272–2279
Endo I, Fukumoto S, Ozono K, Namba N, Tanaka H, Inoue D, Minagawa M, Sugimoto T, Yamauchi M, Michigami T, Matsumoto T (2008) Clinical usefulness of measurement of fibroblast growth factor 23 (FGF23) in hypophosphatemic patients: proposal of diagnostic criteria using FGF23 measurement. Bone (NY) 42:1235–1239
Acknowledgments
We are grateful to the family with ARHR for participation in our study. We thank Dr. Nobuaki Ito and Dr. Seiji Fukumoto of the University of Tokyo for the measurement of serum FGF23 levels and helpful comments.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Koshida, R., Yamaguchi, H., Yamasaki, K. et al. A novel nonsense mutation in the DMP1 gene in a Japanese family with autosomal recessive hypophosphatemic rickets. J Bone Miner Metab 28, 585–590 (2010). https://doi.org/10.1007/s00774-010-0169-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00774-010-0169-0