
Abstract
Metrological comparability as well as reliability of ion activity results measured with ion-selective electrodes (ISE) was investigated within the framework of an interlaboratory comparison between eight partners from national metrology institutes and expert laboratories. Two electrolyte solutions containing the clinically most relevant ions sodium, potassium, magnesium, calcium and chloride having ion activities near the physiological range served as samples. The calibration of the measurement set-ups of the participants was carried out using gravimetrically prepared aqueous electrolyte solutions. The ion activities of these calibration standards were calculated by means of the semi-empirical Pitzer model. The measurement uncertainty of the measurement results was calculated according to the guide to the expression of uncertainty in measurement, GUM. Based on a new scale for ion activities traceable to the SI system of units, comparability and reliability of ISE measurement results of clinically relevant ions is realised.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The concept of traceability of measurement results is mandatory when comparability and reliability of the results is required, especially in the field of clinical chemistry [1–4].
The in vitro diagnostic directive (IV DD, 98/79/EC, Appendix I, 3.) claims that the traceability of values assigned to calibrators and/or control materials must be assured through available reference measurement procedures and/or available reference materials of a higher order [5, 6]. In biological systems mainly electrostatic interactions between ions and non-electrostatic interactions between ions and neutral molecules take place. These interactions lead to a decrease of the effective ion concentration or ion activity, respectively. Therefore, the ion activity is the physiologically more meaningful parameter as ions involved in interactions or bound to other constituents cause a reduction of the number of free ions. The molality-based ion activity a i (unit: mol kg−1) is defined as the product of the molality m i (molar amount of the analyte divided by the mass of the solvent in kg) and the ion activity coefficient γ i (dimensionless) according to equation (1):
In clinical chemistry, the preferably used activity is defined as the product of the amount-of-substance concentration (unit: mol l−1) and the ion activity coefficient (dimensionless).
In order to determine ion activities, ion-selective electrodes (ISE) in potentiometric measurement systems are widely used particularly in clinical chemistry.
These ISE hold ion-selective membranes containing neutral molecules that are specially designed to allow ion detection with high selectivity. Ideally, the measured potential E of the ISE against a reference electrode with a constant electrode potential can be described by the Nikolskij–Eisenman equation (2) [7], which is an extended version of the well-known Nernstian equation,
where the index i specifies the analyte ion and j the interfering ion, E 0 i is the sum of all constant potential differences, E j is the diffusion potential arising at the liquid junction between the measurement solution and the inner electrolyte of the reference electrode, R is the gas constant, T is the absolute temperature, z is the ionic charge number, F is the Faraday constant, a i is the ion activity of analyte ion i, K ij is the selectivity coefficient and m 0 is the reference molality (m 0 = 1 mol kg−1).
Some of the commercially available ion-selective membranes provide very good selectivity to the relative analyte ion, so that the product in the Nikolskij–Eisenman equation containing the interfering ions can be neglected. However, in the case of, e.g., the Mg-selective membrane, the interference term of the Nikolskij–Eisenman equation may be accounted for. Nevertheless, a simple linear model is valid as long as the activities of the interfering ions are supposed to be at a low and constant level. In this case, equation (2) simplifies to equation (3)
where E m is the measured potential.
The differential quotient of equation (3) is the practical slope of the calibration line and usually slightly different from the theoretical value of the Nernstian slope 59.16 mV at 25 °C. The temperature dependence of the Nernstian slope must not be accounted for, as calibration and measurement are carried out at the same temperature.
Due to the linear relation between the measured potential and the logarithm of the activity described above, the activity of an analyte ion of almost any sample can be measured after previous calibration of an ISE set-up with calibration standards of known ion activity. The ion activities in the calibration solutions can be calculated by applying the semi-empirical Pitzer approach, which is based on the well-known Debye–Hückel theory taking into account further interaction terms. Compared to the Debye–Hückel theory, which only takes the dependence of the activity from the ionic strength into account, the Pitzer approach also comprises terms describing among others ion–ion interactions at high ionic strength. Details on the theory and the calculations of ion activities using the Pitzer equation are presented elsewhere [8–10]. A detailed description on the mixed electrolyte containing four chloride salts is given in the Electronic Supplementary Material (ESM) of the paper presented here.
In clinical chemistry, ISE measurements of ion activities of physiological relevant ions are a standard method. For the measurement of sodium and potassium, the International Federation for Clinical Chemistry and Laboratory Medicine (IFCC) recommends to bring the ISE measurement results of ultrafiltrated serum samples into conformity with flame atomic emission spectroscopy measurement results [11].
For the measurement of calcium in serum, the IFCC recommends an ISE set-up on the basis of a calibration with aqueous solutions that have known concentration of ionised calcium and a fixed ionic strength of 0.16 mol kg−1 [12].
Similarly for magnesium, the IFCC recommends measuring and reporting ionised magnesium as a substance concentration relative to the substance concentration of magnesium in the primary aqueous calibrants with concentrations of magnesium, sodium and calcium chloride near the physiological ionic strength of 0.16 mol kg−1 [13].
Today measurements by highly automated instruments—that offer values for up to 200 parameters in a very short time—use diluted serum samples as undiluted samples may still pose handling problems. However, diluted serum samples only yield concentrations but not activities. Fixed ion activity coefficients are defined to report values for the so-called ionised constituents (Na+, K+, Mg2+, Ca2+, Cl−). The activity values are only identical to the values for the ionised constituents if the assumed and fixed ion activity coefficient applied is by chance identical to the one of the unknown sample involved. The information on activity is lost via the dilution of the sample and only total concentration values can be reported.
For historical reasons as well as to avoid confusion, these values for the ionised constituents are converted into concentrations using conversion factors, which are only valid for predefined physiological conditions. In case of, e.g., trauma or renal failure, these factors are not suitable, leading to diagnostic errors that may result in severe consequences for the patient. Furthermore, ion concentration results are apparatus dependent causing further lack of reliability of measurement results in clinical chemistry, particularly in point of care diagnostics [14].
Additionally suitable, mixed, certified standards with known traceability for activity values have been missing up to now for the calibration of the measurement equipment.
The introduction of ion activities in addition to the concentration of an analyte would give a benefit to the clinical chemistry community. Medical staff, including medical doctors, that would be educated to distinguish between both quantities, could still use concentration values at the beginning while getting used to activities and later on use the activity as the physiologically more meaningful parameter. To avoid confusion during this period of transition, all reported values should be unambiguously stated as concentrations or activities. The reader is reminded that in its beginning pH was based on concentration of the hydrated proton and only later on activity was recognised to be more adequate.
Here, we present a new scale for single ion activities, whose values are traced back to the SI system of units and are therefore comparable independent of the apparatus used to determine them. This scale is appropriate to be adopted in clinical chemistry for the purpose of unambiguous diagnosis. Furthermore, it helps to establish higher order reference standards and procedures for ion activity determination to be possibly used in the medical diagnostic area of the future [5].
The scale presented here is applied to pure aqueous sample solutions containing no proteins, as the Pitzer theory does not take protein interactions with ions into account. Nevertheless, the new activity scale is assumed to be applicable to protein containing sample solutions such as blood serum, as no effect of proteins on the ISE measurement is expected if care is taken both on the design and on the composition of the reference electrode [15]. Proteins certainly influence activity but they are not necessary as constituents of calibration solutions whose ion activities are known through the semi-empiric Pitzer approach. The ion-selective electrode will sense ion activity in any case.
Experimental
Uncertainty of activities based on Pitzer’s equation
The Kenneth Sanborn Pitzer’s semi-empirical ion interaction approach was used to evaluate single ion activity coefficients on the molality scale. Complex reference solutions were prepared gravimetrically composed of four high purity chloride salts (NaCl, KCl, MgCl2·6H2O, CaCl2·2H2O) of known stoichiometry and purity that served as activity calibrators for the ion-selective electrode system [16, 17]. Figure 5 of the ESM gives an impression of the number of influence parameters involved in Pitzer ion activity value evaluation for a cation in a complex electrolyte mixture.
In the ESM, the equations of this virial approach are given including the Pitzer ion interaction parameters. Günther Meinrath discussed the role of the Pitzer coefficients uncertainty on ion activities in 2002 just at the time when the new IUPAC pH definition was set-up [18].
A program was written in the Python language using the freeware Metas.Unclib. This program allowed all measured or estimated uncertainty components of the ion activity coefficient to be included [19]. The influence parameters for the Pitzer equations are outlined in the ESM.
The calculated relative expanded (coverage factor k = 2, which represents a 95 % confidence interval) uncertainties U r for the Pitzer ion activity coefficients were less than 0.5 %.
New activity scale
Two steps were used to construct the activity scale. First, electrolyte solutions were prepared gravimetrically from salts with known compositions, and the content of the ions in these solutions were calculated on the basis of the gravimetric data. Second, the ion activities and their associated uncertainties were calculated as described in the previous section.
Electrolyte solutions with known activities were used as standards for the calibration of an ISE set-up. Finally, the potentiometric response of each ion-selective electrode with an ion-selective membrane was measured against the reference electrode. The different potential signals were registered with each membrane and for each solution. The steady state potential signal E was used. These signals were plotted in the graph E versus the logarithm of the corresponding ion activity of each standard solution. The ion activity in samples with unknown content and/or activity were evaluated using the analysis function. Figures 1 and 2 illustrate the concept of an activity scale and how it is established.

General representation of the different steps involved in the determination of an activity scale by potentiometric measurements of electrolyte reference solutions

Schematic representation of the traceability chain for the determination of the activity scale of reference standard solutions
The activity scale presented here should allow comparison of the measured ion activities independent from the utilised measurement device, as the activities are traceable to the SI system of units and therefore comparable among each other. This was checked in an interlaboratory comparison, which is described in the following section.
Interlaboratory comparison
The interlaboratory comparison (EURAMET 1165) was a task of the joint research project T2J10 TRACEBIOACTIVITY of the European Association of National Metrology Institutes (EURAMET) [21]. This project aimed at building up a scale for chemical activity of clinically relevant ions that is consistent with the already existing and accepted activity scale for pH.

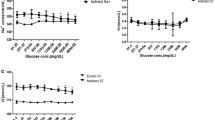
Summary of the activity values and their associated uncertainties for a sodium, b potassium, c chloride, d magnesium and e calcium determined by the partners in the interlaboratory comparison by ISE measurement of solution 1 (I) and solution 2 (II) and evaluation with equation (3) and the reference values (solid lines) together with the range of the associated expanded (k = 2) uncertainties (indicated by dashed lines) at 20 °C
The ISE comparison study was carried out among eight partners (national metrology institutes as well as expert laboratories) in order to assess the reliability and comparability of ion activity measurement results and to validate the activity measurement procedure, developed in the project.

Summary of the molalitiy values and their associated uncertainties determined by participant 1 and participant 7 in the interlaboratory comparison by IC measurement for a sodium, b potassium, c chloride, d magnesium and e calcium of solution 1 (I) and solution 2 (II) and evaluation with equation (3) and the reference values (solid lines) together with the ranges of their associated expanded (k = 2) uncertainties (indicated by dashed lines) at 20 °C
Furthermore, this study was organised in order to compare the measurement values and their assigned combined measurement uncertainties, for the evaluation of the metrological performance of the developed measurement systems and procedures and finally helps for the mutual recognition of calibration and measurement data.
Two solutions containing sodium, potassium, magnesium, calcium and chloride, whose activities are in the molality range depicted in Table 1, were prepared by participant 8 and served as the sample solutions. This molality range—except of chloride—covers the physiological range. The chloride content was somewhat higher than found in normal human serum. The two solutions had different ionic molalities, so that a possible influence of these differences on the behaviour of the ion-selective membranes and furthermore on the measurement uncertainty can be detected. The solutions were gravimetrically prepared from four well characterised high purity chlorides of sodium, potassium, magnesium and calcium, which were purchased from MV Laboratories Inc., Frenchtown (NJ, USA) and fully characterised later on by laboratories of SMU, PTB and METAS.
The characterisation of these salts was carried out by means of coulometric chloride determination in a prepared solution at SMU. These chloride data are used as the reference values as their traceability chain is shortest and the evaluated uncertainties are lowest. The values from three national metrology institutes SMU, PTB and METAS were in agreement. Cation and chloride content are consistent. Also the sum of quantified trace impurities agrees with the chloride content, considering water as an impurity for the alkali salts only. The water content of solid NaCl and solid KCl was measured coulometrically by Karl Fischer titration. The trace elemental contents of the main impurities were measured by inductively coupled plasma mass spectrometry (ICP-MS) whereas ion chromatography was used to measure the main trace anions.
The main purity characteristics of the solid high purity chlorides of sodium, potassium, magnesium and calcium are given in Table 2.
The ion activities of these sample solutions were calculated on the basis of their molalities with the semi-empirical model developed by K. S. Pitzer, which is well accepted and tested and is in agreement with the accepted pH scale [4]. The calculation of the single ion activity coefficients is not only possible for the standard temperature 25 °C as Pitzer’s equation takes the temperature dependence of the activity coefficients into account. Pitzer coefficients, which are temperature dependent, are used as temperature functions (cf. ESM). For practical reasons, the measurements were carried out at 20 °C, but not at physiological temperature (37 °C). Accordingly, the ion activity coefficients calculations were carried out for 20 °C.
The expanded uncertainties of these ion activity coefficients were determined to be about 0.5 % as described above. ISE measurements on the two sample solutions were carried out consistently at 20 °C using a home-designed ISE measurement device. A possible design of such an ISE measurement system can be found in the literature [3]. The devices were calibrated using at least three ion activity standards, which were gravimetrically prepared at each participating institute. The activities in the calibration standards were calculated as mentioned above for the sample solutions.
Furthermore, ion chromatographic measurements were carried out by two interlaboratory comparison participants in order to determine the ion molalities present in the two sample solutions.
Reproducibility of the ion activity as well as of the molality measurements was checked and taken into account in the uncertainty budget. It is clear that the evaluation of the activity coefficients is dependent on all uncertainties involved in the preparation of the solution, including the purity values. Each of the activity coefficients is composed of around 50 parameters and the purity of the reference salts is one of the most prominent influences. In the gravimetric preparation of the salt solutions, we only have about ten influence parameters and also here the uncertainty of the purity value adds prominently to the combined measurement uncertainty of the molality.
The uncertainties of both the ion activity as well as molality results were calculated according to the guide to the expression of uncertainty in measurement (GUM) [1].
Results and discussion
Tables 3 and 4 contain the reference values of the ion activities and the measured ion activities of the sample solutions 1 and 2 together with their associated expanded (coverage factor k = 2) uncertainties U and relative expanded uncertainties U r as well as the degree of equivalence d i , defined as the difference between reference values and measured values for all interlaboratory comparison participants (see Fig. 3 for illustration). Furthermore, the reference values and measured values of the molalities are presented for two interlaboratory comparison participants (see Fig. 4 for illustration).
Ion activities determined by ISE
Visual inspection of the activity measurement results strongly hinted at the presence of at least one outlying result. Therefore, the measurement data were checked for outliers by use of Dixon’s test [22]. The test showed that the activity values determined by participant 6 are outliers except for the potassium result of solution 1 and the calcium result for solution 2. After disregard of the measurement values of participant 6, no outliers remain.
Moreover, the remaining data were checked for mutual consistency by use of a common consistency check as described in the guideline of the Consultative Committee for Amount of Substance—Metrology in Chemistry (CCQM) [2].
Even after deletion of the outliers, the data are mutually inconsistent among the participants except for chloride in solution 1 and for sodium and chloride in solution 2. The reason for this is either a too high scatter of the measurement results or an underestimation of the measurement uncertainties. As some of the results exhibit an uncertainty close to the uncertainty of the reference value, obviously not all uncertainty contributions have been taken into account. Hence, underestimation of the measurement uncertainties is the most likely explanation for the mutual inconsistency of the measurement results.
There are several influences, which may alter the measured potential and hence contribute to the measurement uncertainty.
-
(a)
One uncertainty contribution comes from the limited selectivity of the ion-selective membranes. In the presence of interfering ions, an increase of the measured potential, and therefore, an overestimation of the ion activity may be observed. For example in contrast to solution 1, for solution 2 some of the determined ion activities of Ca and Mg are larger than the reference value. This could be due to the fact, that in solution 2 larger amounts of interfering sodium ions are present. However, this effect was not observed by all institutes. Furthermore, due to the low selectivity of the Mg membrane against Ca2+, the Mg activity determination should have been affected by the presence of Ca2+ ions significantly more than the K+ activity determination. But, this effect could not be observed from the measurement results. Hence, there is no evidence for a contribution of the limited selectivity of the ion-selective membranes to the measurement uncertainty.
-
(b)
Further influences, which may vary the measured potential are, e.g., the geometry of the measurement cell, the fabrication process of the membranes (thickness, ionophore distribution), the conditioning of the membranes (the type of ions present in the conditioning solution, ion concentrations in the conditioning solution, duration of conditioning), the way of fixation of the membranes, the concentration of the inner electrolyte of the ion-selective electrode and the kind of reference electrode used. However, all these effects are the same for all measured solutions and should therefore be eliminated by the calibration process.
-
(c)
Besides the contribution from the measurement reproducibility and the evaluation of Pitzer’s activity coefficients, probably the most important contribution to the measurement uncertainty comes from the liquid-junction potential. A liquid-junction potential develops at the interface of two liquids with different ionic strengths and/or ionic compositions. For example at the interface between the internal solution of the reference electrode and the measurement solution a liquid-junction potential develops. This liquid-junction potential is different for different measurement solutions. Hence, the contribution of the liquid-junction potential to the measurement uncertainty cannot be eliminated with the calibration procedure and therefore has to be taken into account. This correction is the so-called residual-junction potential that is the difference in junction potentials between the one of the calibration solutions and the one of the measurement solutions [12, 23–25].
Henderson calculations give 0.38 mV as the maximum residual liquid-junction potential between two solutions with lowest and highest ionic content according to Table 1; [26]. In order to minimise the uncertainty originating from the residual liquid-junction potential, calibration solutions should be used, which have a similar ionic composition as the sample. This was recommended to the participants in preparation of the outstanding interlaboratory comparison.
The contribution originating from the residual liquid-junction potential has obviously not been taken into account by all institutes. This is the most probable explanation for the inconsistency of some experimental results.
Molalities determined by ion chromatography
Additionally participant 1 and participant 7 determined molalities by use of ion chromatography.
In case of the singly charged ions sodium, potassium and chloride, participant 1 and participant 7 agree well both with each other and with the reference value within the limits of measurement uncertainty for both solutions.
However, in case of the double-charged ions calcium and magnesium, the molalities, measured by participant 1 and participant 7, agree neither with each other nor with the reference value, except for the magnesium molality determined by participant 1, whose values agree well with the reference value within the limits of measurement uncertainty.
The agreement of the measurement values with the reference value is better in case of sodium and chloride as for the other ions. This is the cause of the higher concentrations of sodium and chloride with its larger IC peaks. Thus, for divalent ions, the uncertainty is most probably underestimated.
Overall determination of both, activities or molalities for sodium and chloride, gave better agreement with the reference value than for calcium, magnesium and potassium. This is, among other things, due to the fact that gravimetric preparation of the sample solutions as well as the calibration standards is more uncertain and susceptible to bias in case of calcium and magnesium, as in this case liquids had to be weighed in, which is technically more difficult than weighing solids. The highly hygroscopic chlorides of magnesium and calcium demanded weighing in a controlled atmosphere and the measurement of the exact content by non-gravimetric means. Furthermore, the selectivity of the membrane for the magnesium ions and to a lesser extent for calcium ions is smaller. The fact that potassium gave worse results than sodium and chloride remains unclear.
Conclusions
A new activity scale for the physiologically relevant ions sodium, potassium, magnesium, calcium and chloride has been established. This was achieved by calculation of the ion activities by Pitzer’s equation and determination of the measurement uncertainties of these ion activities according to the GUM. The new activity scale allows comparison of ion activity measurement results independent from the utilised measurement system.
The suitability of the activity scale was subsequently tested in an interlaboratory comparison between eight partners.
The interlaboratory comparison gave satisfactory results for the monovalent ions sodium and chloride, but less satisfactory results for potassium and for the divalent ions calcium and magnesium.
The residual liquid-junction potential was identified as one possible source of uncertainty. If the composition of the measurement solution is unknown, the contribution of the liquid-junction potential to the measurement uncertainty can at least be minimised by use of suitable calibration standard compositions. This was probably not practiced by all participants.
To sum up, an interlaboratory comparison proved for the first time that, at least for sodium and chloride, traceable measurement results can be achieved using ion-selective electrodes. Traceability is a key prerequisite for comparable and reliable ion activity results and therefore a prerequisite for safe and efficient measurements in the clinical laboratory.
In principle, the concept of a new ion activity scale presented here is adaptable to protein-containing samples such as blood serum as already mentioned in the introduction. Performance of a further interlaboratory comparison, using protein-containing samples, would therefore be promising.
References
ISO/IEC (2008) Guide to the expression of uncertainty in measurement (2008), Guide 98-3, Geneva, Switzerland
CCQM (2010) Guidance note: estimation of a consensus KCRV and associated degrees of equivalence
Berdat D, Andres H, Wunderli S (2009) Development of suitable ISE measurement procedures for SI-traceable chemical activity determination. Chimia 63(10):670–677
Buck RP, Rondinini S, Baucke FGK, Brett CMA, Camoes MF, Covington AK, Milton MJT, Mussini T, Naumann R, Pratt KW, Spitzer P, Wilson GS (2002) Measurement of pH. Definition, standards, and procedures (IUPAC Recommendations 2002). Pure Appl Chem 74(11):2169–2200
Communities E Directive 98/79/EC of the European Parliament and the Council on in vitro diagnostic medical devices (1998)
(BIPM) BIdPeM. http://www.bipm.org/jctlm/
Nikolskij BP (1937) Fiz Khim 10:1041
Pitzer KS (1973) Thermodynamics of electrolytes. I. Theoretical basis and general equation. J Phys Chem 77:268–277
Pitzer KS (1974) Thermodynamics of electrolytes. III. Activity and osmotic coefficients for 2-2 electrolytes. J Solution Chem 3:539–546
Pitzer KS, Mayorga G (1973) Thermodynamics of electrolytes. I I. Activity and osmotic coefficients for strong electrolytes with one or both ions univalent. J Phys Chem 77:2300–2308
Burnett RW, Covington AK (2000) Recommendations for measurement of and conventions for reporting sodium and potassium by ion-selective electrodes in undiluted serum, plasma or whole blood. Clin Chem Lab Med 38(10):1065–1071
Burnett RW, Christiansen TF (2000) IFCC recommended reference method for the determination of the substance concentration of ionized calcium in undiluted serum, plasma or whole blood. Clin Chem Lab Med 38(12):1301–1314
Rayana MCB, Burnett RW, Covington AK, D′Orazio P, Fogh-Andersen N, Jacobs E, Külpmann WR, Kuwa K, Larsson L, Lewenstam A, Maas AHJ, Mager G, Naskalski JHJ, Okorodudu AO, Ritter C, John AS (2005) Guidelines for sampling, measuring and reporting ionized magnesium in undiluted serum, plasma or blood. Clin Chem Lab Med 43(5):564–569
Hristova EN, Cecco S, Niemela JE, Rehak NN, Elin RJ (1995) Analyzer-dependent differences in results for ionized calcium, ionized magnesium, sodium and pH. Clin Chem 41:1649–1653
Payne RB, Buckley BM, Rawson KM (1991) Protein interference with ion-selective electrode measurement depends on reference electrode composition and design. Ann Clin Biochem 28:68–72
Pitzer KS (1991) Activity coefficients in electrolyte solutions. CRC Press, Boca Raton
Harvie CE, Møller N, Weare JH (1984) The prediction of mineral solubilities in natural waters: the Na-K-Mg-Ca-H-CI-SO4-OH-HCO3-CO3-CO2-H2O system to high ionic strengths at 25°C. Geochim Cosmochim Acta 48:723–751
Meinrath G (2002) Extended traceability of pH: an evaluation of the role of Pitzer′s equations. Anal Bioannal Chem 374:796–805
Zeier M, Hoffmann J, Wollensack M (2012) Metas. UncLib-a measurement uncertainty calculator for advanced problems. Metrologia 49:809–815
Euramet (2011) Ion activity measurement of Na+, K+, Ca2+, Mg2+ and Cl− in water based mixed electrolyte samples. http://www.euramet.org/index.php?id=tc-projects&tx_nneurametctcp_projects[project]=934&tx_nneurametctcp_projects[action]=show&tx_nneurametctcp_projects[controller]=Project&cHash=30b048374708c8e751609b2460dc9016
Dean RB, Dixon DJ (1951) Simplified statistics for small number of observations. Anal Chem 23(4):636–638
Bates JB, Chu YT, Stribling WT (1988) Surface topography and impedance of metal-electrolyte interfaces. Phys Rev Lett 60(7):627–630
Winkelmann JW, Merritt C, Scott WJ, Kumar A, Baum G (1984) Effects of residual liquid junction potential in direct potentiometry of potassium. Clin Chem 30:482–484
Kadis R, Leito I (2010) Evaluation of the residual liquid junction potential contribution to the uncertainty in pH measurement: a case study on low ionic strength natural waters. Analytica Chimica Acta 664(2):129–135
Henderson P (1907) Zur thermodynamik der flüssigkeitsketten. Z Phys Chem 59:118–128
Acknowledgments
This work has been carried out in the framework of Work Package 3 of the EURAMET Joint Research Project T2/JRP10 “Traceable measurements for biospecies and ion activity in clinical chemistry”. The research within this EURAMET joint research project receives funding from the European Community’s Seventh Framework Programme, ERA-NET Plus, under grant agreement no. 217257.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Bastkowski, F., Spitzer, P., Eberhardt, R. et al. Pitzer ion activities in mixed electrolytes for calibration of ion-selective electrodes used in clinical chemistry. Accred Qual Assur 18, 469–479 (2013). https://doi.org/10.1007/s00769-013-1016-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00769-013-1016-5