
Abstract
Graphene–amino acid interaction is gaining significance mainly based on its possible biomedicine applications. The density functional theory (DFT) calculation and molecular dynamics simulation (MD) are applied to obtain a comprehensive understanding of the adsorption mechanism of three kinds of amino acids, namely, alanine (Ala), glycine (Gly), and valine (Val) over the surface of graphene and functionalized graphene nanosheets. In this study, several analyses such as solvation energy, adsorption energy, intermolecular distances, and charge properties are used to explore the adsorption behavior of amino acid on the nanosheets. The calculated adsorption energies show that the interaction of amino acids with functionalized graphene is greater than the pristine graphene. Regarding DFT computations, the adsorption of Val on the graphene about − 10 kJ/mol is stronger than Gly and Ala. Meanwhile, it is found that the geometrical parameters and electronic properties of graphene change drastically upon functionalization, and the formation of hydrogen bonds between –COOH functional group and amino acids enhances the adsorption energy about 12–30%. To obtain a deeper comprehension of the interaction nature, the atoms in molecules (AIM) and the natural bond orbital (NBO) studies have been performed. Furthermore, the MD simulations are employed to assess the dynamic properties of our designed systems. The results from the present study demonstrate that the movement of the amino acids into the carriers is spontaneous and forms stable complexes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biomolecules in conjunction with nanomaterials including nanoparticles, nanotubes, nanowires, and nanosheets provide new platforms with unique properties that have the synergistic effect of host and guest molecules (Zhang et al. 2013; Emanet et al. 2015).
Among different types of nanomaterials, graphene nanosheet (GNS) has drawn increasing attention of researchers based on its outstanding electronic structure showing high mobility of electron and a remarkable capability to interact with different types of molecules (Bolotin et al. 2008; Neto et al. 2009; Kemp et al. 2013). Furthermore, the graphene is enabled to pass across the biological membrane and has a large surface area which leads to attracting more attention. In several studies, its combination with various biological molecules such as nucleobases (Mirzaei et al. 2012; Vovusha and Sanyal 2015), amino acids (Cazorla 2010; Mallakpour et al. 2017), and different organic structures (Wang et al. 2014b; Zhou and Zhang 2015) has been investigated.
However, based on this fact that the GNS is a single layer of carbon atoms with a hexagonal structure, it is highly hydrophobic, and as a result, it is hardly dispersed in biological systems. For this reason, surface functionalization or modification is required to enhance the graphene solubility and its capability in biological systems. Regarding this, the GNS surface can be modified by the variety of functional groups, including epoxy, carbonyl, and hydroxyl groups to improve the adsorption feature (Yang et al. 2012; Zhao and Liu 2014).
Consequently, the graphene in the functionalized form is utilized as interesting drug delivery systems due to their capability to adsorb high concentrations of drugs (Pulskamp et al. 2007; Ali-Boucetta et al. 2013). In the physiological environment, the high surface area of two-dimensional (2D) materials reveals new horizons for obtaining relatively stronger non-covalent interactions through effective hydrogen bonding and π−π stacking with biomolecules. However, an important prior step in observing physiological reaction of 2D materials is to find out the interaction with biomolecules such as amino acids. It is clearly obvious that deficiencies in even a single vital amino acid could change immunity and enhance disease susceptibility (Hashimoto et al. 2012). Moreover, it has been proposed that the adsorption behavior of tiny molecules like amino acids could be applied to optimize the quantitative–structure activity relationships for prediction of nanomaterials interaction generally with macro-biomolecules including proteins (Xia et al. 2010). In consequence, understanding the interactions between amino acids and 2D materials is crucial to manipulate the developing medical applications.
Furthermore, while pristine graphene 2D nanosheets may show toxicity, graphene decorated with proteins has been found to be less harmful (Ge et al. 2011; Chong et al. 2015). More findings about the formation of protein–graphene complexes can also provoke new trends in bio-based applications of graphene, such as manufacturing developed and effective biosensors, modified drug delivery systems, and protein separation technologies (Huang et al. 2011; Yue et al. 2011; Karunwi et al. 2013). Hence, to increase our capability to design more bio-based complexes, an ability in the prediction of the strength of adsorption and stability of protein structure over the graphene surface is critical. In this regard, several theoretical investigations have been performed to observe the conformational alterations of proteins adsorbed on graphene using a wide variety of theoretical chemistry tools (Zuo et al. 2011, 2017; Penna et al. 2015; Sengupta et al. 2015; Hughes and Walsh 2018; Dasetty et al. 2019). On the other hand, among several types of biomolecules, amino acids perform a crucial role in the living organisms as building blocks of proteins and enzymes. The amino acids can be subdivided according to their properties, dictated by the functional groups they possess. Broadly, they are divided by charge, hydrophobicity, and polarity. These properties influence the way they interact with surrounding amino acids in polypeptides and proteins and, consequently, impact protein 3D structure and properties. Amino acids are good candidates as the suitable promoter for making GNS applicable attention in several experimental and computational researches in (bio) sensors, biomaterials, biomedicine, catalysts, and other fields of science (Popov 2004; Lacerda et al. 2006; Yang et al. 2006; Harrison and Atala 2007; Kang et al. 2009). These studies indicated that interactions in the form of non-covalent, including H-bonding and X–π (X = OH, NH, CH, etc.), are the cause of the nucleobase–GNS and amino acid–GNS complex stabilization (Chen et al. 2003; Kang 2005; Roman et al. 2006; Gowtham et al. 2007; Varghese et al. 2009; Cazorla 2010; Umadevi and Sastry 2011). Furthermore, it is found that proteins, nucleobases, and small molecules can interact with functionalized graphene effectively (Chen et al. 2003; Kang 2005; Roman et al. 2006; Gowtham et al. 2007; Varghese et al. 2009; Umadevi and Sastry 2011; Cazorla et al. 2012). For instance, Pan and coworkers used molecular dynamic (MD) simulations to study the adsorption behavior of glutamine acids and glycine on the surface of the hydroxyapatite crystalline in the form of rod and plate structure (Pan et al. 2007). Also, density functional theory (DFT) calculation is used by Zhiani to explore adsorption of several amino acids such as asparagine, arginine, cysteine, and histidine on the graphene and boron nitride (BN) nanosheet. The results revealed that the adsorption process of the amino acids on the BN nanosheet and the graphene surface accompanied by the energy release (Zhiani 2017). Moreover, it is shown that no covalent bonded is formed between the amino acids and the substrate surfaces. Podila et al. experimentally investigated the nature of interactions and charge transfer direction between aromatic amino acids and (2D) materials including graphene, graphene oxide, and boron nitride. The results obtained from these calculations are indicated the graphene and GO strongly interact with the aromatic amino acids through π−π stacking and H-bonding (Mallineni et al. 2016).
In another study, Qi et al. employed DFT calculation and MD simulation to find the adsorption geometries and energies, electronic band structures, and the adsorption dynamics of l-leucine/graphene complex. Their obtained results showed the stability of adsorption energy and confirmed that the electronic structure of graphene can be dominated by the adsorption direction of l-leucine (Qin et al. 2010). They observed that interactions between l-leucine and the graphene were in the form of physisorption and l-leucine prefers to give parallel configuration to the graphene surface.
To the best of our knowledge, there is a lack of information about the adsorption mechanism of some of the amino acids with graphene and functionalized graphene, and moreover, nature and unavoidable features of their interactions remain obscure, which has slowed down the pace of the biological and biomedical applications of graphene.
Alanine (Ala), glycine (Gly), and valine (Val) are important and simple three amino acids in humans. They act as precursors for several key metabolites of low molecular weight such as creatine. Shortage of these three basic amino acids in small quantities is not harmful to health, but severe shortage may lead to failure of the immune response, low growth, abnormal nutrient metabolism, and undesirable health effects (Ganji 2009). Therefore, they are considered conditionally essential amino acids for humans and other mammals to enhance good growth.
In this work, the DFT calculations are performed to obtain the adsorption energies, geometries, and electronic properties of amino acid/graphene systems. Furthermore, the effect of functionalization of GNS with a carboxyl group (–COOH) on the amino acid–GNS interactions in the water solvent and gas phase is explored. Moreover, the MD simulation is used to gain deep insight from the dynamic and diffusion properties of amino acid adsorption on GNS and functionalized GNS in a biological environment. We believe that the obtained results from this work can be used in future pharmacological studies.
Computational procedure
Quantum mechanics calculations
Theoretical calculations were accomplished by the DFT method to analyze the interactions between several types of amino acids with graphene (GNS) and functionalized graphene nanosheet (f-GNS). The optimized structure of amino acids obtained from the QM calculations and graphene structure built using the Nanotube Modeler package. It is to be noted that the employed model of both the nanosheets as well as three amino acids considered is neutrally charged. The GNS model contained 54 carbon atoms and the bonds at the carrier edges were saturated with H atoms. Three carboxyl groups were replaced with hydrogen atoms at the edge of GNS to construct the f-GNS model (C57H18O6).
The initial geometries of the pure graphene, functionalized graphene nanosheet, and the investigated complexes are performed in both vacuum and aqueous phase utilizing the Gaussian package (version 03) (Frisch et al. 2008) at ωB97XD with the standard 6–31G** basis set. The ωB97XD functional integrates the long-range (LC), short-range, as well as including explicit empirical dispersion corrections (DFT-D) (Chai and Head-Gordon 2008a, b).
As known that aqua media play a considerable role in the human body, therefore, it can be chosen as a solvent to specify the interaction of various types of amino acids with graphene and functionalized graphene nanosheet to mimic the biological environment. Tomasi's polarized continuum (PCM) model (Miertuš et al. 1981; Lipparini and Mennucci 2016) has been employed for the solvent effect of water calculation.
To assess the compatibility of the nanosheets towards interacting amino acids, adsorption energy (Eads) which is the main parameter in both gas and solvent, has been calculated using Eq. (1):
where Ecomplex, Eadsorbent, and Eamino acid are energies of the studied configurations, the graphene and functionalized graphene, and the free amino acids, respectively. Besides, the basis set superposition error (BSSE) is computed by means of the counterpoise method (Boys and Bernardi 1970) to remove the basis function overlap effects.
The atoms in molecule (AIM) (Yoosefian 2017; Kamel et al. 2019a) is employed to probe the electron transfer between nanosheets and amino acids using AIM2000 program (Biegler-König 2000). Also, using Espinosa method (Espinosa et al. 1999), the hydrogen-bond energies (EHB = 1/2Vb) have been calculated. To realizing the orbital interactions and delocalization of charge in the investigated structures, the Natural Bond Orbital (NBO) analysis is performed (Reed et al. 1992).
Molecular dynamics simulation
The MD simulation is applied to scrutinize the dynamic properties and adsorption behavior of the investigated complexes. Six simulation boxes are designed in which one of the investigated amino acids (i.e., Gly, Ala, or Val) is placed above the carrier (GNS or f-GNS). The simulation boxes are considered with dimensions 4 nm × 4 nm × 4 nm. All of the force field parameters for the nanosheets and the amino acids are selected from the Charmm36 force field. The TIP3P (Jorgensen et al. 1983) water molecules are added to the simulation boxes and then, to provide a correct biological environment and neutralize the systems, Na+ and Cl− ions are replaced with some waters. All of MD productions are ran under periodic boundary conditions for 45 ns using the GROMACS package (version 5.1.4) (Abraham et al. 2015). The temperature and pressure are maintained at 310 K and 1 bar by using the V-rescale thermostat and Berendsen barostat, respectively (Berendsen et al. 1984; Bussi et al. 2007). The Particle-Mesh-Ewald (PME) procedure is employed to assess the long-range electrostatic interactions. The interactions with non-bonded are computed with a cutoff in the range of 1.2 nm (Darden). Moreover, the VMD software is utilized for trajectory visualization.
Computational results
DFT calculation results
The theoretical results in this work are obtained by means of quantum mechanics calculations and MD simulations. First, we accomplished the quantum analysis to survey the adsorption of amino acid molecules onto the surfaces of graphene and functionalized graphene. In this section, the adsorption energies, the structural and electronic characteristics, as well as topological parameters and orbital hybridization for the interaction of amino acid molecules with the nanosheets are scrutinized in the vacuum and aqueous phases.
Geometrical analysis
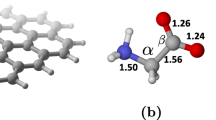
Figure 1 exhibits the optimized structures of then nanosheets (GNS and f-GNS) and amino acids (Gly, Ala, and Val). All of the investigated amino acids have three active sites, i.e., the carboxyl (COOH), the amino (NH2), and the side chain (R-group), differing only in the R-group or side chain they have. Different orientations of amino acids to the graphene and the functionalized graphene nanosheet are explored utilizing the Spartan package (Shao et al. 2006) to gain the most stable structure of each complex. Then, the full optimization of these most stable complexes has been performed at \(\omega\) B97XD functional level. The optimized structures of the designed systems are depicted in Fig. 2.

Optimized structures of the Gly, Ala, Val, GNS, and f-GNS at the wB97XD /6-31G** level

Optimized structures of the amino acid/GNS and amino acid/f-GNS complexes at the wB97XD /6-31G** level
Table 1 illustrates the computed energies for the investigated structures in both the gas and solution phase. By the same token, the equilibrium distances and the structural parameters of the amino acid molecules, GNS, f-GNS, and the considered complexes are presented in Tables T2 and S1, supplementary material. For the hydrogen-saturated GNS, the average C–C distance of the benzene rings is 1.41 Å, which is in line with the obtained results from previous studies (Shahabi and Raissi 2017, 2018).
Exploring the adsorption behavior of the amino acid molecules onto the pristine graphene surface shows that guest molecules are located on top of the host at distances about 0.3 nm. The range of the adsorption energy for the considered systems demonstrates that the amino acid molecules can be physically adsorbed on the graphene nanosheet surface in both the gas phase and the water solution. This type of physisorption effect is in line with weak interactions that extensively exist and play a vital part in biological systems (Ganji 2009; Qin et al. 2010).
Moreover, (Zhiani 2017) reported that adsorption energy for the complex of the alanine amino acid and GNS is − 34.18 kJ/mol. The adsorption energy for different amino acid–carrier complexes is listed in Table S4 (Qin et al. 2010; He and Zhou 2014; Wang et al. 2014a; Zhiani 2017). In this work, the calculated Eads has a good accordance with the reported value for interactions of amino acid molecules and the graphene in the previous studies (Qin et al. 2010; He and Zhou 2014; Wang et al. 2014a; Zhiani 2017). It is found that the adsorption process has physical nature and amino acid molecules preferred to parallelly arrange on the graphene surface. Furthermore, it is observed GNS and f-GNS have a good potential for interaction with Val and other amino acid molecules.
Alternatively, to elucidate the influence of the functional group on the strength of intermolecular interactions, we have employed three functional groups of COOH on the GNS nanosheet. Comparing the values of adsorption energy from Table 1, it is clearly evident that, upon the functionalization of the GNS nanosheet surface, the adsorption energies have significantly improved. Our theoretical findings indicate that the adsorption energy values for Gly/f-GNS, Ala/f-GNS, and Val/f-GNS in the gas phase (water solution) are − 37.63 (− 87.72) kJ/mol, − 44.07 (− 96.25) kJ/mol, and − 64.07 (− 115.54) kJ/mol, respectively. As can be observed in Table 1, the adsorption energy of amino acids is augmented by functionalization. It seems that the formation of hydrogen bond (HB) between the amino acid molecules and the carboxylic functional groups results in more stability of amino acid/f-GNS complexes. It can be concluded that the carboxylic functional groups not only can decline the toxicity of graphene but also rises amino acid molecules adsorption on the carrier surface. With respect to the acquired results from Table 1, it is observed that, by applying a solvent effect, the adsorption energy of the complexes is drastically changed (Castro et al. 2015; Kaur et al. 2015).
The solvation energy parameter is calculated to assay the solubility of the selected structures in water media and the gained results are listed in Table 1. Negative solvation energy values illustrate that the solvating process is spontaneous, which is evidence of the stability of the studied systems in the aqueous environment. The large solvation energy of the amino acid adsorbed f-GNS surface is confirmed its high performance as a nanocarrier in the biological systems (Singla et al. 2016).
Clearly, based on the obtained results, the Val amino acid, an amino acid having aliphatic side-chains with a branch, in all the cases has a more tendency in approaching the adsorbent surfaces in comparison to the other amino acid molecules. On the other hand, the adsorption energy is maximum for a valine (the largest amino acid); it can be interpreted that, based on fundamentals of van der Waal’s (vdW) forces, the obtained results are directly corresponding to the mass and size of the interacted molecules. Consistent with previous studies, the weight factors used to minimize the energies obtained by molecular and quantum mechanics simulations (Robertson et al. 2015; Dasetty et al. 2019). For adsorption of valine on the functionalized graphene, two H bondings are formed at the top of the nanosheet edge; one is O75···H100–O82 which O75 atom of the carboxyl group of f-GNS functions as the HB acceptor and the other one is O76–H77…O83 which hydroxyl (O76–H77) group of the –COOH functional group behaves as the HB donor. The formation of hydrogen bonds strengthens the interaction of the amino acids with the f-GNS.
Also, it is well known that the strength of interactions can be reflected in the geometrical parameters of the considered complex. Principally, stronger interactions well correlate with shorter intermolecular distances (Kamel et al. 2017). Regarding shorter intermolecular distances between amino acids and functionalized nanosheet in comparison to pristine nanosheet, as a result, it is found that the f-GNS remarkably behave as a preferable surface for the adsorption of the amino acids (Table 2). Also, as shown in this table, it can be concluded that in going from the gaseous phase to the water solution, the intermolecular equilibrium distance values reduce. Otherwise speaking, the intermolecular interactions between amino acid molecules and f-GNS are stronger in the water solution.
As can be noticed from Table 2, the intermolecular Of-GNS…Hamino acid (H bond between amino acids and functionalized graphene) distances in the most stable amino acid/f-GNS complexes are evidently shorter than the Hf-GNS…Oamino acid; subsequently, in these complexes, the former H bonds are much stronger than the latter case. The shortest Of-GNS…Hamino acid distance is observed in the Val amino acid; hence, the result is a stronger interaction. This consequence in line with the highest negative adsorption energy value for Val/f-GNS complex. The physisorption with short adsorption distance well supports the preservation of the biological activity of Val amino acid on graphene in addition to transfer performance.
Moreover, the structure parameters of the adsorbed amino acids on GNS and f-GNS are also summarized in Table S1, supplementary material. We observe that the bond length, as the geometrical parameter of the system, almost remains unchanged in comparison to the isolated structures.
Furthermore, for guaranteeing structural stability, the vibration frequency for the interaction between adsorbent surfaces and amino acid molecules is evaluated. Table S2, supplementary material, exhibits detailed information of the vibrational frequencies computed for the studied complexes. It can be inferred that, owing to the formation of intermolecular HB between f-GNS and amino acid molecules, the stretching mode of the O–H band of amino acid moieties shifts from 3800 cm−1 in isolated molecules to the lower frequencies at 3200 cm−1 in studied complexes (see Table S2, supplementary material). Furthermore, the obtained results display that the vibration frequencies of the N–H, C=O, and C–H bands are slightly changed from the amino acid molecules to the considered complexes, implying the existence of weak interaction between amino acid molecules and nanosheets.
Atoms in molecule analysis
To perceive further comprehension into the nature of the intermolecular interactions of adsorbed amino acids on GNS and f-GNS surfaces, the topological properties of electron density are computed by the AIM theory (Yoosefian et al. 2016; Kamel et al. 2019b). Tables 3 and S3, supplementary material, encompass the electron density (ρBCP), its Laplacian (∇2ρBCP) at bond critical point, the kinetic electron energy density (GBCP), the local potential electron energy density (VBCP), and the total electron energy density (HBCP) values for the considered systems in the gas phase and the water solution.
Rozas et al. (2000) stated that the intermolecular interactions can be distinguished using the values and signs of ∇2ρBCP and HBCP. The positive value of the ∇2ρBCP demonstrates a reduction of charge density in the intermolecular region between amino acid molecules and GNS which confirms the presence of a non-covalent interaction (see Tables 3 and S3, supplementary material). Molecular graphs of interactions between the adsorbent surfaces and the amino acids are also displayed in Fig. 3. Molecular graphs obtained for the optimized complexes illustrate critical points and their related bond paths. It can be found from the obtained results that the highest number of intermolecular BCPs belongs to Val amino acid in both phases. Furthermore, our theoretical results show that amino acid/f-GNS complexes have low ρBCP and negative values of ∇2ρBCP and HBCP in all complexes which prove the formation of strong intermolecular HBs in both gas phase and water solution (Hasanzade and Raissi 2019).

Molecular graph of interaction amino acids with GNS and f-GNS obtained from the DFT calculation
The lower values of the ρBCP and ∇2ρBCP parameters for interaction of the amino acid with GNS in comparison to corresponding values for f-GNS exhibit the weaker intermolecular interactions in amino acid/GNS complex (see Tables 3 and S3, supplementary material). This consequence is in accordance with smaller adsorption energy values presented in Table 1.
Depending on the outcomes of Tables 3 and S3, supplementary material, it is quite obvious that electron density values, in the regions of Of-GNS …Hamino acid HB interaction, are more than the other interactions in both phases. These conclusions are well supported by the shorter interaction distances (see Table 2) and can be related to the fact that the complex with the shortest intermolecular distance is associated with the greatest ρBCP at interatomic BCP.
Closer inspection of the results from Tables 3 and S3, supplementary material, reveals that the highest EHB value belongs to the conjunction between the hydrogen atom of valine molecule and the oxygen atom of the functionalized graphene nanosheet, which is major evidence of being the strongest HB bonds in this structure. Also, these outcomes are in good agreement with the smaller HB distances (see Table 2). Figure 4 presents the relation between topological parameters versus EHB in the investigated structures. The correlation coefficients are very near to the unit 1.

Correlation graphic of the topological parameters and EHB energy values
Population analysis from NBO perspective
Electron transfer is one key index in the adsorption mechanism of an adsorbate on an adsorbent (Zaboli et al. 2020). For evaluating charge transfer in the studied molecular systems as well as for studying intermolecular orbital overlaps upon complexion, the NBO method is performed and computed results are tabulated in Table 4. The NBO analysis reveals that in the considered complexes during the intermolecular interactions, charge is transferred from the amino acid molecule to nanosheet. This result is expected, because the number of heteroatoms in the amino acid is more than nanosheet. As clearly can be observed in this table, the second-order perturbation energy (E(2)) values for the different amino acid/GNS complexes vary between 0.08 to 0.11 and 0.08 to 0.15 kcal/mol in the gas phase and the water solution, respectively. It is found that in the case of the Val amino acid, the large stabilization effect is acquired because of the strong orbital interactions between the sigma bonding orbital of the O74–C80 bond in the valine molecule and the anti-bonding (π*) of the C23–C36 bond in graphene nanosheet with the values of 0.11 kcal/mol and 0.15 kcal/mol in the gas phase and water solution, respectively.
With changing the adsorbent from the graphene to the functionalized graphene, it is observed that the charge transfer becomes more intense. The lone pair of O83 of the amino acid molecule (donor role) overlaps with the LP* C74 of the COOH functional group of graphene (as acceptor) at the Val/f-GNS complex. This overlap leads to electronic charge transfer from LPO83 to LP*C74 with the E(2) values of 0.97 and 2.16 kcal/mol in the gas phase and the water solution, respectively (see Table 4). The acquired results of the NBO method authenticate that the charge transfers between amino acids and f-GNS are higher than their values for GNS. Besides, it is found that the most prominent stabilization energy for Val/f-GNS complex is in line with the above results (see Tables 2 and 4).
Also, the higher values of E(2) energy after amino acid molecule interaction with f-GNS indicates a greater proportion of its interaction. These findings are also associated with the large adsorption energies of amino acid/f-GNS models in comparison with the amino acid/GNS configurations. It is quite obvious that higher charge transfer energies are related to greater absolute values of the adsorption energy (see Tables 1 and 4).
Our findings clearly show that there is a prominent linear correlation between EHB and E(2) energy in the investigated functionalized complexes as indicated in Fig. S1, supplementary material. It is quite evident that by enhancing the HB energy, the stronger intermolecular interactions can be observed. Furthermore, the charge transfer of charge between the two ingredients will be extended.
Thermodynamic parameters
To study the stability of the considered amino acid/f-GNS (GNS) complexes, the thermodynamic parameters of the selected systems are computed in two phases and the acquired findings are presented in Table 1. The negative values of the standard enthalpy (ΔH) and Gibbs free (ΔG) energies illustrate that the amino acid adsorption process on the GNS and f-GNS surfaces is exothermic and spontaneous in both phases. According to Table 1, it is evident that the formation of the complex of Val amino acid with the GNS and f-GNS is thermodynamically more stable than the other investigated amino acids.
The consequences of our computations reveal a linear behavior between Eads and the ΔH of the structures with the correlation coefficient of 0.947 (see Fig. S2, supplementary material). Overall, it may be concluded that ΔH is a beneficial parameter to the survey of the adsorption energy.
Molecular dynamics simulation results
In this section, the dynamic and adsorption behavior of amino acid molecules onto GNS and f-GNS will be studied.
To visualize the adsorption process, the final orientations of the amino acid molecules on the carrier surfaces are extracted from the production trajectories (Fig. S3). In MD simulation, we allow all of the components to move freely without any restraints. At first, the amino acid molecules are distributed randomly around the nanosheets. It should be noted that amino acid molecules are located approximately 2 nm away from the carriers to minimize the effect of starting orientations. During MD simulation, the distance between some of the amino acid molecules and the nanosheets is decreased, and finally, the amino acid spontaneously adsorbed on the surface of carriers. This finding has a good agreement with DFT calculations, which indicated the formation of amino acid–carrier complexes due to the negative adsorption energy is exergonic.
For evaluating the distribution of the amino acids around the carriers, the radial distribution function (RDF) is calculated and depicted in Fig. 5. The most probable distance for finding amino acid molecules around the carrier surfaces is cf. 0.4–0.8 nm. This result approves that amino acid molecules tend to physisorbed on the nanosheets. Since the function g(r) for the amino acid/f-GNS system started in the shorter distances than the GNS system, it is more probable to find amino acid around of the functionalized system at closer distances. This result may be related to the formation of HB between the functionalization groups and the amino acid molecules. Also, these distances have a good correlation with the obtained intermolecular distances from the DFT calculation as well as with ranges of amino acid–carrier interaction distances in previous investigations (Ganji 2009; Singla et al. 2016; Dasetty et al. 2019). A comparison of the maximum peaks of amino acid interactions in two different systems displays that the adsorption behavior changes with changing surface chemistry.

Radial distribution functions (RDF) of the amino acid molecules with GNS and f-GNS
To acquire better comprehension of the diffusion characteristics of amino acid molecules and the adsorption process kinetics of systems, several parameters can be assessed. Figure 6 exhibits the vdW interaction energy of the amino acid molecules with the nanosheets based on the function of time. Obviously, it is found that, by the adsorption of amino acid molecules on graphene and functionalized graphene nanosheet surface, a reduction in the vdW energy is obtained. This behavior indicated that the amino acid molecules approach the carrier surfaces spontaneously and nearly adsorbed on the surface of nanosheets after 0.5 ns. The gained consequences can be proved by the steep decrease of the vdW energies after this time.

Van der Waals interactions of the amino acid molecules with GNS and f-GNS
Also, Table 5 shows the assessment of the energy between the nanosheets and the amino acid molecules. The negative values of vdW and electrostatic (elec) energies for the interaction of amino acid molecules with the nanosheets approve that the adsorption process is exergonic and spontaneous. These findings have a good agreement with the results of DFT adsorption energy, and show that the GNS and f-GNS are good carriers for the adsorption of amino acids. We also have surveyed the average electrostatic interactions during the adsorption mechanism for considered structures. It can be understood that, for interaction of the amino acids with the GNS, the contribution of vdW energy is more than elec energy. This result is expected due to the hydrophobic nature of the graphene nanosheet. On the other hand, by the addition of functional groups containing oxygen to the graphene surface, the impact of electrostatic interaction in the adsorption process is raised, whereas the contribution of vdW interaction is decreased. This increase is due to the formation of a hydrogen bond between the amino acid molecule and the functional group. Generally, amino acid adsorption on the functionalized system is stronger than the pristine system to be in good agreement with the results of the DFT calculations.
Close inspection of Table 5 illustrates that the electrostatic interaction has a high impact on the adsorption of Ala molecule on functionalized graphene (− 141.37 kJ/mol). This finding may be related to the formation of strong HB between amino acid molecules and f-GNS (as mentioned in "3.1.2 Atoms in molecule analysis" section). In other words, the formation of more number and stronger hydrogen bonds in the Ala/f-GNS system leads to an increase in the electrostatic contribution of the amino acids-nanosheets binding. Analyzing the vdW interactions of the studied systems showed that the interaction Val with GNS is more favorable than the other amino acid–nanosheet pair. This behavior can be related to the significant contribution of \(\sigma\)–\(\pi\) interactions in vdW interactions. Overall, in line with DFT results, the interaction of amino acids with f-GNS is stronger than GNS.
Furthermore, hydrogen-bond pattern, which was calculated using the gmx hbond module of GROMACS, was used to obtain more insight into the hydrogen-bond changes during MD simulations. The number of calculated H bonds in the studied systems is depicted in Figure S4. It is worthwhile to note that the number of hydrogen bonds between the Ala molecule and the functionalized graphene is more than other amino acid molecules. Therefore, it can be concluded that the electrostatic interaction and hydrogen bond have significant roles in the promotion of non-covalent association between the Ala molecule and f-GNS.
The mean-square displacements (MSDs) of the amino acid molecules in the selected systems are depicted in Fig. 7. The MSD is calculated based on the following equation:

The mean-square displacements (MSDs) of the amino acid molecules with GNS and f-GNS
Where \(r_{{{\text{AA}}}} \left( t \right) - {\text{r}}_{{{\text{AA}}}} \left( 0 \right)\) is the distance traveled by center of mass (COM) of the particle AA over some time interval of length \(\Delta t\).
The self-diffusion coefficient (Di) for amino acids and nanosheets gained from the slope of MSD in MD calculations and tabulated in Table 5. The obtained values of the diffusion coefficient in the considered systems show that the diffusion coefficient for Val amino acid in the GNS and f-GNS systems is the least. This fact can be attributed to the stronger binding and more molecular weight of this amino acid to the carriers. Also, based on the acquired results in the DFT part, it can be stated that the number of HB can prevent the displacement of amino acid molecules.
Conclusion
In the current study, the interaction between three amino acids with graphene and functionalized graphene is comprehensively inspected using DFT calculations and MD simulations in both the gas phase and water solvent. DFT calculations confirmed the energetic stability of adsorption model, and revealed that electronic structure of graphene can be controlled by the adsorption direction of DFT calculations on amino acid/GNS and amino acid/f-GNS systems show that interactions between amino acid molecules with the GNS and f-GNS are in the type of physisorption and confirmed the energetic stability of adsorption model. The values of solvation and adsorption energies confirm that the functionalized graphene nanosheet is more appropriate as an adsorbent surface for amino acids in comparison to the graphene surface. Also, the most important factor in the stability of amino acid/f-GNS complexes is hydrogen-bond interactions. All the quantum mechanics computations demonstrate that, among the three kinds of amino acids considered in this survey, Val forms the most stable complexes with both surfaces. Remarkably, these observations indicate that there is a direct correlation between the size and the binding strength of three different amino acids. Moreover, to examine the dynamic of amino acid adsorption on GNS and f-GNS surfaces, MD simulations are accomplished. It is observed that the vdW interaction is the main force in the adsorption of amino acid molecules on graphene. While by adding oxygen-containing to the surface of nanosheet, the role of electrostatic interaction is raised. The present work supports that graphene and functionalized graphene are efficient substrates for non-covalently binding of amino acids. Besides the complication of real proteins, on the other hand, all of them are constructed from amino acids. Therefore, the obtained results in this paper can be used for evaluating the stability proteins on the GNS and f-GNS.
References
Abraham MJ, Murtola T, Schulz R et al (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25
Ali-Boucetta H, Bitounis D, Raveendran-Nair R et al (2013) Purified graphene oxide dispersions lack in vitro cytotoxicity and in vivo pathogenicity. Adv Healthc Mater 2:433–441
Berendsen HJC, van Postma JPM, van Gunsteren WF et al (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690
Biegler-König F (2000) University of applied sciences: Bielefeld
Bolotin KI, Sikes KJ, Jiang Z et al (2008) Ultrahigh electron mobility in suspended graphene. Solid State Commun 146:351–355
Boys SF, de Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126:14101
Castro L, Kirillov E, Miserque O et al (2015) Are solvent and dispersion effects crucial in olefin polymerization DFT calculations? Some insights from propylene coordination and insertion reactions with group 3 and 4 metallocenes. ACS Catal 5:416–425
Cazorla C (2010) Ab initio study of the binding of collagen amino acids to graphene and A-doped (A= H, Ca) graphene. Thin Solid Films 518:6951–6961
Cazorla C, Rojas-Cervellera V, Rovira C (2012) Calcium-based functionalization of carbon nanostructures for peptide immobilization in aqueous media. J Mater Chem 22:19684–19693
Chai J-D, Head-Gordon M (2008a) Systematic optimization of long-range corrected hybrid density functionals. J Chem Phys 128:84106
Chai J-D, Head-Gordon M (2008b) Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys 10:6615–6620
Chen RJ, Bangsaruntip S, Drouvalakis KA et al (2003) Noncovalent functionalization of carbon nanotubes for highly specific electronic biosensors. Proc Natl Acad Sci 100:4984–4989
Chong Y, Ge C, Yang Z et al (2015) Reduced cytotoxicity of graphene nanosheets mediated by blood-protein coating. ACS Nano 9:5713–5724
Darden T, York D, Pedersen L (1993) J J Chem Phys 98:10089
Dasetty S, Barrows JK, Sarupria S (2019) Adsorption of amino acids on graphene: assessment of current force fields. Soft Matter 15:2359–2372
Emanet\cSenÇobandedeÇulha MOZM (2015) Interaction of carbohydrate modified boron nitride nanotubes with living cells. Colloids Surfaces B Biointerfaces 134:440–446
Espinosa E, Souhassou M, Lachekar H, Lecomte C (1999) Topological analysis of the electron density in hydrogen bonds. Acta Crystallogr Sect B Struct Sci 55:563–572
Frisch MJ, Trucks GW, Schlegel Hb, et al (2008) Gaussian 03, revision C. 02
Ganji MD (2009) Density functional theory based treatment of amino acids adsorption on single-walled carbon nanotubes. Diam Relat Mater 18:662–668
Ge C, Du J, Zhao L et al (2011) Binding of blood proteins to carbon nanotubes reduces cytotoxicity. Proc Natl Acad Sci 108:16968–16973
Gowtham S, Scheicher RH, Ahuja R et al (2007) Physisorption of nucleobases on graphene: density-functional calculations. Phys Rev B 76:33401
Harrison BS, Atala A (2007) Carbon nanotube applications for tissue engineering. Biomaterials 28:344–353
Hasanzade Z, Raissi H (2019) Carbon and boron nanotubes as a template material for adsorption of 6-Thioguanine chemotherapeutic: a molecular dynamics and density functional approach. J Biomol Struct Dyn 38:1–11
Hashimoto T, Perlot T, Rehman A et al (2012) ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 487:477–481
He Z, Zhou J (2014) Probing carbon nanotube–amino acid interactions in aqueous solution with molecular dynamics simulations. Carbon N Y 78:500–509
Huang Y, Dong X, Liu Y et al (2011) Graphene-based biosensors for detection of bacteria and their metabolic activities. J Mater Chem 21:12358–12362
Hughes ZE, Walsh TR (2018) Probing nano-patterned peptide self-organisation at the aqueous graphene interface. Nanoscale 10:302–311
Jorgensen WL, Chandrasekhar J, Madura JD et al (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Kamel M, Raissi H, Morsali A (2017) Theoretical study of solvent and co-solvent effects on the interaction of Flutamide anticancer drug with Carbon nanotube as a drug delivery system. J Mol Liq. https://doi.org/10.1016/j.molliq.2017.10.078
Kamel M, Raissi H, Hashemzadeh H, Mohammadifard K (2019a) Understanding the role of hydrogen bonds in destruction of DNA by screening interactions of Flutamide anticancer drug with nucleotides bases: DFT perspective, MD simulation and free energy calculation. Adsorption 26:1–18
Kamel M, Raissi H, Morsali A, Mohammadifard K (2019b) Density functional theory study towards investigating the adsorption properties of the $γ$-Fe 2 O 3 nanoparticles as a nanocarrier for delivery of Flutamide anticancer drug. Adsorption 26:925–939
Kang HS (2005) Theoretical study of binding of metal-doped graphene sheet and carbon nanotubes with dioxin. J Am Chem Soc 127:9839–9843
Kang Y, Liu Y-C, Wang Q et al (2009) On the spontaneous encapsulation of proteins in carbon nanotubes. Biomaterials 30:2807–2815
Karunwi O, Baldwin C, Griesheimer G et al (2013) Molecular dynamics simulations of peptide–swcnt interactions related to enzyme conjugates for biosensors and biofuel cells. Nano Life 3:1343007
Kaur J, Singla P, Goel N (2015) Adsorption of oxazole and isoxazole on BNNT surface: A DFT study. Appl Surf Sci 328:632–640
Kemp KC, Seema H, Saleh M et al (2013) Environmental applications using graphene composites: water remediation and gas adsorption. Nanoscale 5:3149–3171
Lacerda L, Bianco A, Prato M, Kostarelos K (2006) Carbon nanotubes as nanomedicines: from toxicology to pharmacology. Adv Drug Deliv Rev 58:1460–1470
Lipparini F, Mennucci B (2016) Perspective: polarizable continuum models for quantum-mechanical descriptions. J Chem Phys 144:160901
Mallakpour S, Abdolmaleki A, Borandeh S (2017) Fabrication of amino acid-based graphene-zinc oxide (ZnO) hybrid and its application for poly (ester–amide)/graphene-ZnO nanocomposite synthesis. J Thermoplast Compos Mater 30:358–380
Mallineni SSK, Shannahan J, Raghavendra AJ et al (2016) Biomolecular interactions and biological responses of emerging two-dimensional materials and aromatic amino acid complexes. ACS Appl Mater Interfaces 8:16604–16611
Miertuš S, Scrocco E, Tomasi J (1981) Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem Phys 55:117–129
Mirzaei M, Ravi S, Yousefi M (2012) Modifying a graphene layer by a thymine or a uracil nucleobase: DFT studies. Superlattices Microstruct 52:306–311
Neto AHC, Guinea F, Peres NMR et al (2009) The electronic properties of graphene. Rev Mod Phys 81:109
Pan H, Tao J, Xu X, Tang R (2007) Adsorption processes of Gly and Glu amino acids on hydroxyapatite surfaces at the atomic level. Langmuir 23:8972–8981
Penna MJ, Mijajlovic M, Tamerler C, Biggs MJ (2015) Molecular-level understanding of the adsorption mechanism of a graphite-binding peptide at the water/graphite interface. Soft Matter 11:5192–5203
Popov VN (2004) Carbon nanotubes: properties and application. Mater Sci Eng R Rep 43:61–102
Pulskamp K, Wörle-Knirsch JM, Hennrich F et al (2007) Human lung epithelial cells show biphasic oxidative burst after single-walled carbon nanotube contact. Carbon N Y 45:2241–2249
Qin W, Li X, Bian W-W et al (2010) Density functional theory calculations and molecular dynamics simulations of the adsorption of biomolecules on graphene surfaces. Biomaterials 31:1007–1016
Reed AE, Carpenter JE, Wienhold F (1992) NBO version 3.1. Gaussian, Inc, Pittsburgh
Robertson MJ, Tirado-Rives J, Jorgensen WL (2015) Improved peptide and protein torsional energetics with the OPLS-AA force field. J Chem Theory Comput 11:3499–3509
Roman T, Dino WA, Nakanishi H, Kasai H (2006) Amino acid adsorption on single-walled carbon nanotubes. Eur Phys J D-Atomic Mol Opt Plasma Phys 38:117–120
Rozas I, Alkorta I, Elguero J (2000) Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J Am Chem Soc 122:11154–11161
Sengupta B, Gregory WE, Zhu J et al (2015) Influence of carbon nanomaterial defects on the formation of protein corona. RSC Adv 5:82395–82402
Shahabi M, Raissi H (2017) Investigation of the solvent effect, molecular structure, electronic properties and adsorption mechanism of Tegafur anticancer drug on Graphene nanosheet surface as drug delivery system by molecular dynamics simulation and density functional approach. J Incl Phenom Macrocycl Chem 88:159–169
Shahabi M, Raissi H (2018) Screening of the structural, topological, and electronic properties of the functionalized Graphene nanosheets as potential Tegafur anticancer drug carriers using DFT method. J Biomol Struct Dyn 36:2517–2529
Shao Y, Molnar LF, Jung Y et al (2006) Spartan’08, Wavefunction, Inc., Irvine, CA. Phys Chem Chem Phys 8:3172–3191
Singla P, Riyaz M, Singhal S, Goel N (2016) Theoretical study of adsorption of amino acids on graphene and BN sheet in gas and aqueous phase with empirical DFT dispersion correction. Phys Chem Chem Phys 18:5597–5604
Umadevi D, Sastry GN (2011) Quantum mechanical study of physisorption of nucleobases on carbon materials: graphene versus carbon nanotubes. J Phys Chem Lett 2:1572–1576
Varghese N, Mogera U, Govindaraj A et al (2009) Binding of DNA nucleobases and nucleosides with graphene. ChemPhysChem 10:206–210
Vovusha H, Sanyal B (2015) Adsorption of nucleobases on 2D transition-metal dichalcogenides and graphene sheet: a first principles density functional theory study. Rsc Adv 5:67427–67434
Wang M, Guo Y, Wang Q et al (2014a) Density functional theory study of interactions between glycine and TiO2/graphene nanocomposites. Chem Phys Lett 599:86–91
Wang X, Liu B, Lu Q, Qu Q (2014b) Graphene-based materials: fabrication and application for adsorption in analytical chemistry. J Chromatogr A 1362:1–15
Xia X-R, Monteiro-Riviere NA, Riviere JE (2010) An index for characterization of nanomaterials in biological systems. Nat Nanotechnol 5:671–675
Yang M, Yang Y, Yang H et al (2006) Layer-by-layer self-assembled multilayer films of carbon nanotubes and platinum nanoparticles with polyelectrolyte for the fabrication of biosensors. Biomaterials 27:246–255
Yang X, Niu G, Cao X et al (2012) The preparation of functionalized graphene oxide for targeted intracellular delivery of siRNA. J Mater Chem 22:6649–6654
Yoosefian M (2017) Powerful greenhouse gas nitrous oxide adsorption onto intrinsic and Pd doped single walled carbon nanotube. Appl Surf Sci 392:225–230
Yoosefian M, Ansarinik Z, Etminan N (2016) Density functional theory computational study on solvent effect, molecular conformations, energies and intramolecular hydrogen bond strength in different possible nano-conformers of acetaminophen. J Mol Liq 213:115–121
Yue R, Lu Q, Zhou Y (2011) A novel nitrite biosensor based on single-layer graphene nanoplatelet–protein composite film. Biosens Bioelectron 26:4436–4441
Zaboli M, Raissi H, Moghaddam NR, Farzad F (2020) Probing the adsorption and release mechanisms of cytarabine anticancer drug on/from dopamine functionalized graphene oxide as a highly efficient drug delivery system. J Mol Liq 301:112458
Zhang C, Peng Z, Lin J et al (2013) Splitting of a vertical multiwalled carbon nanotube carpet to a graphene nanoribbon carpet and its use in supercapacitors. ACS Nano 7:5151–5159
Zhao X, Liu P (2014) Biocompatible graphene oxide as a folate receptor-targeting drug delivery system for the controlled release of anti-cancer drugs. RSC Adv 4:24232–24239
Zhiani R (2017) Adsorption of various types of amino acids on the graphene and boron-nitride nano-sheet, a DFT-D3 study. Appl Surf Sci 409:35–44
Zhou P-P, Zhang R-Q (2015) Physisorption of benzene derivatives on graphene: critical roles of steric and stereoelectronic effects of the substituent. Phys Chem Chem Phys 17:12185–12193
Zou X, Wei S, Jasensky J et al (2017) Molecular interactions between graphene and biological molecules. J Am Chem Soc 139:1928–1936
Zuo G, Zhou X, Huang Q et al (2011) Adsorption of villin headpiece onto graphene, carbon nanotube, and C60: effect of contacting surface curvatures on binding affinity. J Phys Chem C 115:23323–23328
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling editor: A. G. de Brevern.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kamel, M., Raissi, H., Hashemzadeh, H. et al. Theoretical elucidation of the amino acid interaction with graphene and functionalized graphene nanosheets: insights from DFT calculation and MD simulation. Amino Acids 52, 1465–1478 (2020). https://doi.org/10.1007/s00726-020-02905-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-020-02905-5